
Praca doktorska [PDF]
Zachodniopomorski Uniwersytet Technologiczny w Szczecinie
Wydział Technologii i Inżynierii Chemicznej
Rozprawa doktorska
„Badania porowatych materiałów węglowych otrzymywanych poprzez karbonizację poli(tereftalanu etylenu) w mieszaninie z wybranymi związkami magnezu”
Justyna Zatorska
Promotor:
dr hab. inż. Jacek Przepiórski, prof. ZUT w Szczecinie
Szczecin 2013
Dziękuję:
mojemu promotorowi Panu dr hab. inż. Jackowi Przepiórskiemu, prof. ZUT za poświęcony czas i cenne uwagi w trakcie realizacji niniejszej pracy doktorskiej
całemu zespołowi naukowemu i doktorantom Zakładu Technologii Wody i Inżynierii Środowiska, a w szczególności Panu mgr inż. Adamowi Czyżewskiemu
za życzliwość i okazane wsparcie
Szczególne podziękowania dedykuję mojemu mężowi Wojtkowi,
moim Rodzicom i Dziadkom oraz siostrze Judycie za wsparcie w chwilach zwątpienia i motywację do pracy, bez Was nie dotarłabym tak daleko
2
Spis treści
1. Węgiel aktywny ............................................................................................................ 5
1.1 Ogólna charakterystyka węgli aktywnych.....................................................................5
1.2 Czynniki determinujące strukturę porowatą węgla aktywnego ..................................7
1.2.1 Wpływ natury surowca oraz warunków karbonizacji ...................................... 7
1.2.2 Aktywacja karbonizatu ..................................................................................... 8
1.3 Alternatywne metody otrzymywania porowatych materiałów węglowych ............13
1.4 Przetwarzanie odpadów z poli(tereftalan etylenu) .....................................................17
1.5 Węgle aktywne w selektywnej adsorpcji zanieczyszczeń wody ..............................21
2. Cel i zakres pracy........................................................................................................ 27
3. Charakterystyka stosowanych materiałów.................................................................. 28
3.1 Komercyjny węgiel aktywny CWZ-35 ........................................................................28
3.2 Prekursor węgla – poli(tereftalan etylenu) (PET).......................................................28
3.3 Związki magnezu ............................................................................................................28
3.4 Modelowe zanieczyszczenia organiczne .....................................................................28
4. Preparatyka węgli aktywnych ..................................................................................... 31
4.1. Przygotowanie mieszaniny związku nieorganicznego z PET ..................................31
4.2. Karbonizacja mieszanin związek magnezu/PET .......................................................31
4.3 Wydzielanie materiałów węglowych z otrzymanych karbonizatów ........................32
5. Metody analityczne ..................................................................................................... 34
5.1 Analiza termograwimetryczna (TG).............................................................................34
5.2 Dyfrakcja promieniowania rentgenowskiego (XRD) ................................................34
5.3 Pomiar izoterm adsorpcji/desorpcji azotu w 77 K......................................................35
5.4 Termoprogramowalna desorpcja (TPD) .....................................................................35
5.5 Rentgenowska spektroskopia fotoelektronów (XPS).................................................36
5.6 Spektrofotometria w zakresie nadfioletu i promieniowania widzialnego (UV-Vis)36
5.6.1 Pomiar adsorpcji zanieczyszczeń organicznych z roztworów wodnych na otrzymanych węglach aktywnych ........................................................................... 37
6. Wyniki i dyskusja ....................................................................................................... 38
6.1. Wyznaczenie warunków karbonizacji wyjściowej mieszaniny...............................38
6.1.1 Analiza TG PET ............................................................................................. 38
3
6.1.2 Analiza TG i XRD związków nieorganicznych oraz ich mieszanin z PET po karbonizacji ............................................................................................................. 39
6.2 Charakterystyka otrzymanych materiałów węglowych .............................................44
6.2.1 Pomiar izoterm adsorpcji/desorpcji N2 w 77 K wybranych materiałów węglowych............................................................................................................... 44
6.2.2 Wpływ temperatury karbonizacji wyjściowych mieszanin oraz ich składy, na rozwój porowatości w otrzymanych materiałach węglowych................................. 49
6.2.3 Rozkład objętości porów w wybranych materiałach węglowych .................. 51
6.3 Mechanizm tworzenia porów w otrzymanych materiałach węglowych..................53
6.4 Wpływ domieszek na wydajność tworzenia węgla w układach związek
magnezu/PET .........................................................................................................................61
6.5 Zdolność adsorpcyjna materiałów węglowych względem wybranych zanieczyszczeń organicznych ..............................................................................................64
6.5.1 Usuwanie fenolu z wody ................................................................................ 65
6.5.2 Usuwanie barwników z wody ........................................................................ 68
7. Wnioski ....................................................................................................................... 74
Spis rysunków ................................................................................................................. 75
Spis tabel......................................................................................................................... 78
Streszczenie .................................................................................................................... 79
Abstract ........................................................................................................................... 80
Wykaz literatury cytowanej ............................................................................................ 81
4
1. Węgiel aktywny
1.1 Ogólna charakterystyka węgli aktywnych
Węgle aktywne to amorficzne materiały węglowe charakteryzujące się bardzo silnie rozwiniętą porowatością i powierzchnią wewnętrzną. Odgrywają one m.in. znaczącą rolę w procesach adsorpcyjnego oczyszczania cieczy i gazów ze składników o małej koncentracji. Adsorbenty węglowe są powszechnie stosowane do usuwania z wód barwy, smaku, zanieczyszczeń pochodzenie organicznego i nieorganicznego [1, 2]. Materiały te znalazły także zastosowanie w procesach oczyszczania powietrza z zanieczyszczeń pochodzących z przemysłowych gazów odlotowych [3, 4, 5]. Właściwości węgli aktywnych są w istotny sposób związane z jego budową zależną od natury surowca wyjściowego oraz sposobu i warunków preparatyki.
Produkcja węgli aktywnych oparta jest na różnorodnych materiałach pochodzenia organicznego, takich jak drewno [6; 7], węgle kopalne [8], lignina [9], pestki owoców [10], skorupy orzechów [11] oraz materiały pochodzenia syntetycznego [12, 13]. Preparatyka węgli aktywnych obejmuje karbonizację surowego materiału w atmosferze obojętnej i aktywację otrzymanego karbonizatu. W trakcie pirolizy materiału wyjściowego, większość pierwiastków niewęglowych (tlen, wodór, azot, siarka) jest eliminowana
w postaci produktów gazowych, a atomy węgla pierwiastkowego tworzą warstwy złożone z pierścieni aromatycznych powiązanych nieregularnie, pomiędzy którymi powstają wolne przestrzenie, tworzące strukturę porowatą węgli aktywnych. Produkt karbonizacji ma słabo rozwiniętą strukturę porowatą i z reguły wykazuje niewielką powierzchnię właściwą. Struktura ta rozwijana jest w procesie aktywacji, gdzie przestrzenie między aromatycznymi warstwami oczyszczane są w znacznej mierze z węgla bezpostaciowego. W wyniku procesu aktywacji, karbonizat przekształca się w postać zawierającą możliwie największą ilość przypadkowo rozłożonych porów o różnych kształtach i rozmiarach, których obecność w decydującym stopniu wpływa na zdolność adsorpcyjna materiałów węglowych względem związków o różnych
wielkościach cząsteczek, tak z gazów jak i z cieczy [14].
5
makropory
mezopory
mikropory
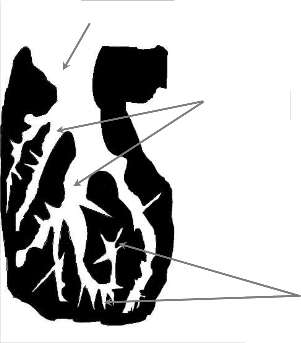
Rysunek 1. Model budowy węgla aktywnego [15]
Węgle aktywne mają pory o rozmiarach zaczynających się poniżej 1 nm i sięgające do kilku tysięcy nanometrów. Klasyfikacja porów według Międzynarodowej Unii Chemii Czystej i Stosowanej (International Union of Pure and Apllied Chemistry, IUPAC), opiera się na ich szerokości, która stanowi odległość między ściankami poru o kształcie szczelinowym lub promieniu poru cylindrycznego [14]. Pory dzieli się na trzy grupy:
Mikropory, <2nm
o Ultramikropory, <0,5nm
o Supermikropory, od 1 do 2nm
Mezopory, od 2 do 50nm
Makropory, >50nm
Efektywność procesu adsorpcyjnego zależy przede wszystkim od parametrów struktury porowatej materiałów węglowych. Na ogół praktyczne zastosowanie węgli aktywnych wymaga, aby te adsorbenty miały dużą zawartość drobnych porów. Proces adsorpcji w mikroporach zachodzi na skutek objętościowego zapełniania. Powierzchnia właściwa mikroporów stanowi nawet do 95% całkowitej powierzchni właściwej węgla aktywnego
i w decydującym stopniu decyduje o pojemności adsorpcyjnej adsorbatu [14]. Mezopory
6
czyli tzw. pory przejściowe charakteryzuje kondensacja kapilarna adsorbentu. Ze względy na rozmiar przyczyniają się one w znaczącym stopniu do adsorpcji adsorbatu o większych rozmiarach cząstek jak na przykład barwiki czy kwasy humusowe. Mezopory działają, jako swoiste kanały, przez które cząsteczki adsorbatu przemieszczają się do wnętrza mikroporów. Natomiast makropory, z reguły nie mają istotnego znaczenia dla procesu adsorpcji na węglu aktywnym. Ma to związek z ich bardzo małym udziałem w powierzchni całkowitej adsorbentu. Pełnią one rolę kanałów
transportowych dla adsorbentu do mikro- i mezoporów [16].
Ze względu na silną zależność pomiędzy strukturą porowatą i efektywnością adsorpcji różnych adsorbatów, dobór adsorbentu, pod względem jego powierzchni właściwej i dominującego rozmiaru porów, powinien być skorelowany z właściwościami usuwanych molekuł.
1.2 Czynniki determinujące strukturę porowatą węgla aktywnego
Struktura porowata materiałów węglowych, a co za tym idzie ich zdolność adsorpcyjna względem zanieczyszczeń jest w dużym stopniu zdeterminowana przez właściwości surowca wyjściowego oraz warunki preparatyki, takich jak parametry karbonizacji oraz rodzaj czynnika wykorzystywanego w procesie aktywacji.
1.2.1 Wpływ natury surowca oraz warunków karbonizacji
Dostosowując rodzaj surowca i parametrów wstępnej obróbki termicznej, można w dość szerokim zakresie wpływać na strukturę porowatą materiału, tj. na stopień rozwinięcia powierzchni i rozkład rozmiarów porów. Surowiec przeznaczony do produkcji węgli aktywnych powinien wykazywać małą zawartość części lotnych, dużą zawartość węgla pierwiastkowego oraz wysoką odporność mechaniczną, tj. na rozkruszenie i ścieranie [17].
Właściwości karbonizatu warunkowane są również przez parametry pirolizy takimi jak: końcowa temperatura karbonizacji, szybkość grzania oraz czas jej trwania. Ważnym czynnikiem jest także atmosfera, w której karbonizacja jest prowadzona. Wysoka temperatura karbonizacji skutkuje zwiększoną ilością uwalnianych lotnych frakcji
i niskocząsteczkowych produktów pirolizy z surowca wyjściowego, a w rezultacie
7
wpływa na końcową wydajność produktu i jego porowatość. W porównaniu z materiałami otrzymanymi w niższej temperaturze [18, 19], materiał węglowy otrzymany w wyższej temperaturze karbonizacji często charakteryzuje się znacznie większą objętością porów. Ponadto, im wyższa temperatura tym głębiej przebiegają procesy pirolizy materiałów, a tym samym powstają materiały charakteryzujące się wyższą wytrzymałością mechaniczną [17]. Szybkość ogrzewania surowca wyjściowego podczas procesu karbonizacji może wpływać także na reaktywność karbonizatu podczas oddziaływania z gazowym aktywatorem. Reaktywność karbonizatów otrzymanych przy dużej szybkości ogrzewania jest większa niż otrzymanych podczas powolnego wzrostu temperatury [20]. W trakcie szybkiego wzrost temperatury, poszczególne etapy rozkładu termicznego prekursora węgla oraz wtórne reakcje produktów pirolizy nakładają się. Z tego powodu powstawanie struktury porowatej w karbonizacie jest trudniejsze do kontrolowania. Szybki wzrost temperatury nie sprzyja uporządkowaniu budowy chemicznej substancji węglowej, a w krótkim okresie wydziela się duża ilość części lotnych. Częstym efektem tego jest powstanie w materiale porów o większych rozmiarach. Powolne ogrzewanie zwęglonego materiału sprzyja tworzeniu się struktury mikroporowatej. Jednakże, zbyt długi czas przetrzymywania materiału w końcowej temperaturze karbonizacji, wpływa znacząco na zmniejszenie się powierzchni właściwej materiału [21]. Atmosfera, w której prowadzi się proces karbonizacji ma znaczący wpływ na rozkład termiczny prekursora węgla oraz przebieg reakcji produktów pirolizy ze stałym karbonizatem. Wysokie przepływy gazu inertnego, generalnie powodują obniżenie wydajności karbonizatu, jednakże jego reaktywność jest większa [17]. Produktem procesu karbonizacji są praktycznie nieaktywne adsorpcyjnie
materiały, o powierzchni właściwej rzędu 20 – 80 m2/g. W celu uzyskania efektywnego
adsorbentu, koniecznym jest otwarcie niedostępnych porów i ogólne zwiększenie ich objętości. W tym celu stosuje się częściowe zgazowanie otrzymanego karbonizatu, czyli tzw. aktywację.
1.2.2 Aktywacja karbonizatu
Tekstura materiału węglowego zaczyna się kształtować już podczas procesu karbonizacji surowca wyjściowego (rozdział 1.2.1). Dalszy rozwój porowatości i powierzchni właściwej materiałów węglowych, jak wspomniano w rozdziale 1.1, jest wynikiem procesu aktywacji, któremu poddawane są produkty karbonizacji
8
prekursorów węgla. Wyróżniamy dwa rodzaje aktywacji fizyczną oraz chemiczną. W przypadku procesu aktywacji fizycznej rozwinięcie struktury porowatej materiału węglowego oparte jest na częściowym zgazowaniu karbonizatu czynnikiem gazowym, najczęściej parą wodną [22], ditlenkiem węgla [23] bądź ich mieszaniną [24], w temperaturze od około 800 °C do 1100 °C [25]. Reakcje chemiczne zachodzące podczas procesu aktywacji ilustrują reakcje 1 – 3:
C + H2 O → CO + H2 (1) C + CO2 → 2CO (2) CO + H2O → CO2 + H2 (3)
Usuwanie amorficznego węgla oraz niejednorodne utlenianie elementarnych kryształów prowadzi w pierwszej fazie aktywacji do powstania nowych porów, czyli do rozwinięcia struktury makroporowatej. W następnych fazach procesu następuje rozszerzenie się istniejących porów, lub powstanie porów o większych wymiarach, przez całkowite wypalenie się ścianek pomiędzy sąsiednimi mikroporami. Prowadzi to do wzrostu objętości makroporów, podczas, gdy objętość mikroporów zmniejsza się. Miarą stopnia aktywacji jest procentowe zmniejszenie się wagi materiału podczas aktywacji, w stosunku do wagi skarbonizowanego produktu wyjściowego, czyli tzw. wypalenia. Gdy stopień wypału osiąga wartość pomiędzy 50 – 75% materiał węglowy
wykazuje zwykle strukturę mieszaną, tzn. zawiera zarówno mikro- jak i mezopory [26].
Aktywację prowadzi się zazwyczaj w atmosferze CO2 lub pary wodnej. Użycie ditlenku węgla, jako czynnika aktywującego w procesie aktywacji umożliwia lepszą kontrolę struktury mikroporowatej materiału węglowego oraz wpływa na jej silniejsze rozwinięcie [27]. Natomiast zastosowanie czynnika aktywującego w postaci pary wodnej umożliwia otrzymanie węgli aktywnych o większej powierzchni właściwej oraz rozbudowanym systemem szerszych mikroporów i mezoporów [28]. Ważnymi czynnikami wpływającym na rozwój struktury porowatej materiałów węglowych są temperatura procesu oraz szybkość przepływu czynnika aktywującego. Dowiedziono, że zwiększenie stopnia przepływu CO2 sprzyja reakcji gazowego czynnika aktywującego z węglem w położeniach aktywnych. Węgle aktywne o maksymalnej powierzchni właściwej (1700 m2/g) zostały otrzymane w temperaturze aktywacji
900 °C, w czasie 240 minut, z przepływem 800 cm3/min [28]. Przeprowadzenie procesu
aktywacji karbonizatu otrzymanego z łupin orzecha kokosowego ditlenkiem węgla
9
w wyższej temperaturze (950 °C) znacznie zwiększyło objętość porów w materiale
węglowym [29].
Wpływem parametrów procesu aktywacji na strukturę porowatą produktu zajmowali się również Qinyan Yue i współpracownicy [30]. W ich pracy surowcem wyjściowym był odpad przemysłu papierniczego, a aktywację parą wodną prowadzono w zakresie temperatur od 700 °C do 850 °C, w czasie od 10 do 150 minut. Badania wykazały, że wraz z podwyższeniem temperatury aktywacji (od 700 °C do 725 °C) zewnętrzna powierzchnia właściwa i całkowita objętość porów uległy istotnemu zwiększeniu. Powyżej temperatury 750 °C powierzchnia BET stopniowo malała, co miało związek z poszerzeniem i zniszczeniem istniejących porów. W temperaturze 800 °C, nastąpiło
wyraźne zwiększenie powierzchni mikroporów oraz nieznaczny wzrost powierzchni właściwej. Efekt ten można przypisać obecności wodoru, który hamuje reakcje pary wodnej z węglem [31]. 30 – 40 minut wskazano jako optymalny czas aktywacji. W tych warunkach otrzymano węgle aktywne o największej powierzchni właściwej, w granicach 270 – 310 m2/g.
Główną zaletą aktywacji fizycznej jest stosunkowo niski koszt wytworzenia jednostkowej ilości węgla aktywnego. Jednakże w trakcie procesu występuje znaczny ubytek masy surowca [32], co znacznie wpływa na obniżenie opłacalności produkcji. Aktywacja chemiczna łączy proces karbonizacji i aktywacji w jeden etap. Polega na reakcji prekursora węgla z czynnikiem aktywującym, takimi jak kwas fosforowy (V) (H3PO4) [33,34], chlorek cynku (II) (ZnCl2) [35, 36] czy wodorotlenek potasu lub sodu
(KOH, NaOH) [37, 38]. L. Hsu i H. Teng [39], zbadali wpływ różnych reagentów
chemicznych (ZnCl2, H3PO4, KOH) oraz temperatury procesu na strukturę porowatą węgli aktywnych otrzymanych z bitumicznego węgla. Ze względu na utlenianie węgla i mechanizm gazyfikacji, proces aktywacji KOH wypłynął w większym stopniu na obniżenie wydajności węglowej, niż w przypadku użycia ZnCl2 czy H3PO4. Bez względu na rodzaj użytego aktywatora, porowatość materiałów węglowych wzrastała wraz z temperaturą karbonizacji. Prekursory węgla aktywowane alkaliami, charakteryzowały się silnie rozwiniętą strukturą mikroporowatą oraz wyższą powierzchnią właściwą przekraczającą nawet 3000 m2/g [39, 40]. Różnicę tę autorzy
przypisują różnemu charakterowi chemicznemu reagentów [41, 42]. H. Younesi i in. [40]
dowiedli, że stosunek reagentów aktywujących względem surowca wyjściowego wpływa na porowatość produktu i pozwala kontrolować strukturę porowatą produktu końcowego. K. Kierzak i in. [43] opisali możliwości wpływania na właściwości węgli
10
aktywnych otrzymywanych z różnych surowców pochodzenia węglowego w procesie aktywacji wodorotlenkiem potasu i sodu. Potwierdzono, że najsilniejszy wpływ na rozwinięcie struktury porowatej węgla aktywnego miała temperatura wstępnej obróbki termicznej surowca (stopień karbonizacji) oraz stosunek wagowy KOH/surowiec w mieszaninie wyjściowej. Zwiększenie zawartości KOH znacząco wpłynęło na wzrost objętości porów i powierzchni właściwej. Wykazano również, że wraz ze wzrostem stopienia karbonizacji surowca węglowego malała powierzchnia właściwa i objętość porów, jak również wzrastał udział mikroporów w materiale węglowym.
Proces aktywacji chemicznej, wiąże się z koniecznością oddzielenia od materiału węglowego stałych reagentów i produktów ich rozkładu od oraz ich regenerację lub utylizację [44]. Ze względu na problemy związane z korozją oraz ograniczenia związane z odzyskiem ZnCl2, wykorzystanie tego czynnika aktywującego zostało praktycznie zarzucone [45]. W przypadku aktywacji kwasem ortofosforowym, temperatura procesu
jest dość niska (do 500 °C), jednakże jego odzysk wiąże się z wieloetapowym procesem ekstrakcji [45]. Powoduje to znaczny wzrost kosztów produkcji węgli aktywnych. Aktywacja materiałów wyjściowych alkaliami pozwala na uzyskanie materiałów węglowych o charakterze mikroporowatym, o szczególnie wysokiej powierzchni właściwej i wąskim rozrzucie wymiarów porów. Jednakże, rozkład wymiaru porów materiałów węglowych powstających w wyniku aktywacji chemicznej, jest uzależniony od stosunku ilościowego aktywatora do prekursora węgla. W przypadku niskiego
stosunku impregnacji, objętość mikroporów jest relatywnie niska. W celu otrzymania materiałów wysoce mikroporowatych, koniecznością jest użycie większej ilości KOH w stosunku do prekursora materiału węglowego [16].
Istnieje także możliwość połączenia aktywacji fizycznej i chemicznej w jeden proces [46]. Metoda ta skutkuje otrzymaniem materiałów węglowych o dobrze rozwiniętej powierzchni właściwej [47, 48]. N. V. Sych i in. otrzymali wysoce porowate węgle aktywne poprzez zastosowanie procesu aktywacji parą wodną, poprzedzonego
impregnacją prekursora węgla kwasem siarkowym.
11![]()

Prekursor węgla
Obróbka wstępna
Impregnacja kwasem siarkowym
Przemycie w odą
destylowaną
Suszenie w 110 °C
Połączony proces aktywacji
(chemiczna + para wodna)
![]()
Wysoce porowaty materiał węglowy
Rysunek 2. Proponowany proces technologiczny otrzymywania wysoce porowatych
materiałów [49]
Proponowane połączenie obu rodzajów aktywacji, może stać się alternatywą dla powszechnie stosowanej aktywacji fizycznej. Powyższa metoda skraca relatywnie czas aktywacji oraz znacznie obniża temperaturę procesu [49].
Rozwój szeroko rozumianej technologii stwarza nowe możliwości oraz wymagania, co do parametrów porowatych materiałów węglowych. Zarówno procesy aktywacji fizycznej i chemicznej mają duże znaczenie praktyczne, jednakże obwarowane są one pewnymi niedogodnościami. Proces aktywacji fizycznej nie ponosi za sobą wysokich kosztów, w wytworzeniu jednostkowej ilości materiału węglowego, przez co węgiel aktywny jest powszechnie stosowany w różnych gałęziach przemysłu. Jednakże, aktywacja fizyczna związana jest ze znacznym ubytkiem masy materiału. Zaletą aktywacji chemicznej jest, co prawda ominięcie wstępnego etapu karbonizacji surowca oraz otrzymanie węgla aktywnego z większą wydajnością. Aczkolwiek powszechnie stosowane czynniki aktywujące muszą być oddzielone od otrzymanej jednostki węglowej. Ponosi to za sobą znaczny wzrost kosztów procesu. Z tego powodu podejmowane są liczne próby nad otrzymywaniem węgli aktywnych, często
o kontrolowanej strukturze porowatej, metodami alternatywnymi.
12
1.3 Alternatywne metody otrzymywania porowatych materiałów węglowych
Zapotrzebowanie na węgle aktywne o specyficznych i oczekiwanych właściwościach ciągle wzrasta. Nowoczesny przemysł często wymaga węgli aktywnych o ściśle określonych objętościach porów, określonym zakresie wielkości porów lub odpowiedniej powierzchni właściwej. Przykładem wysoce mikroporowatych materiałów węglowych o wysokiej powierzchni właściwej są węglowe sita molekularne (CMS, ang. Carbon Molecular Sieves). W ich budowie można wyróżnić szczeliny (przewężenia) i komory (szersze pory). Wąskie szczeliny odseparowują większe cząsteczki, uniemożliwiając ich wnikanie do wnętrza komór. Łączna objętość komór
określa pojemność adsorpcyjną sita molekularnego [14]. Węglowe sita molekularne
można otrzymać z węgli aktywnych w procesie ich wtórnej obróbki. Generalnie wyróżnia się dwie metody preparowania sit molekularnych z węgli aktywnych: poprzez kontrolę procesu pirolizy węglowego prekursora lub zmniejszenie średnicy już istniejących porów poprzez osadzanie atomów węgla lub związku na powierzchni węgla aktywnego, w celu zablokowania wejścia do szerszych porów [50]. M. A. Ahamd i in. [50] modyfikowali zdolność adsorpcyjną materiałów węglowych w stosunku do
gazów przez osadzanie na nich węgla, wytworzonego w wyniku pirolizy par benzenu. Piroliza benzenu prowadzona nad powierzchnią węgla aktywnego prowadzi do osadzania depozytu węglowego na wejściach do porów, zmniejszając ich średnicę. Powyższa procedura pozwoliła na otrzymanie węglowych sit molekularnych o średnicy porów między 0,35 – 0,38 nm, wykazujących wysoką selektywność do rozdzielnia układów O2/N2, CO2/CH4. Stwierdzono, że wydłużenie czasu osadzania węgla powstałego przez termiczny rozkład par benzenu [51] powoduje zmniejszenie rozmiaru wejść do porów (mniejsza objętość mikroporów). Autorzy sugerują, że może to być przyczyną zatykania porów, z powodu ich nadmiernego wypełnienia depozytem
węglowym. B. N. Kuznetsov i in. otrzymali węglowe sita molekularne z hydrolitycznej ligniny, stosując proces karbonizacji i aktywacji parą wodną [52]. Szybkość ogrzewania w procesie karbonizacji okazała się głównym parametrem regulującym strukturę mikroporowatą materiałów. Niska szybkość ogrzewania (0,2 – 2,2 °C/min) w zakresie temperatur od 400 °C do 700 °C, promowała tworzenia się mikroporów ze średnim rozmiarem około 0,56 – 0,58 nm. Wraz ze wzrostem szybkości grzania zwiększył się średni rozmiaru mikroporów (do 0,70 – 0,78 nm). Proces aktywacji otrzymanych
materiałów przyczynił się do zwiększenia objętości mikroporów do 0,30 – 0,35 cm3/g,
13
z szerokością od 0,60 nm do 0,66 nm. Materiały węglowe otrzymane z ligniny wykazały właściwości sit molekularnych, zdolnych do rozdziału mieszaniny He/CH4. A. Lizzo i M. Rostam-Abadi [53] spreparowali węglowe sita molekularne stosując, jako
surowiec wyjściowy niskouwęglony węgiel kamienny. Surowiec ten poddano karbonizacji w różnych temperaturach oraz aktywacji KOH. Strukturę porów wybranych materiałów węglowych modyfikowano poprzez deponowanie węgla pochodzącego z pirolizy metanu. Otrzymane materiały wykazały duża powierzchnię właściwą, a rozkład wielkości mikroporów wykazał obecność porów o wymiarach około 0,4 nm. Materiały węglowe wykazały dobre zdolności skutecznego rozdzielania mieszanin O2/N2, CO2/CH4, CH4/H2.
Prowadzonych jest wiele badań mających na celu otrzymanie węgli aktywnych
o kontrolowanej strukturze porowatej, z pominięciem oddzielnie prowadzonego etapu aktywacji fizycznej. W ostatnio stosowanych metodach produkcji porowatych węgli o ściśle zdefiniowanej strukturze porowatej używa się przestrzennych materiałów nieorganicznych jak zeolity lub silikażele [54, 55, 56]. Stanowią one matrycę dla prekursora węgla, która poddawana jest odpowiedniej obróbce termicznej. Usuwanie matrycy stanowi końcowy etap procesu. Jednak, należy wziąć pod uwagę, że usunięcie matrycy wiąże się często z zastosowaniem korozyjnych kwasów, jak np. kwasu
fluorowodorowego. Poza tym, niesie to za sobą niską wydajność węglową procesu. Ostatnio proponowaną techniką umożliwiającą otrzymanie porowatych materiałów węglowych, o ściśle kontrolowanej strukturze jest tzw. metoda „blending”. Polega ona na zmieszaniu ze sobą fizycznie lub chemicznie najczęściej dwóch polimerów. Mieszaninę poddaje się procesowi karbonizacji, podczas którego jeden z polimerów pod wpływem temperatury tworzy matrycę węglową, a drugi ulega rozkładowi do produktów gazowych, pozostawiając pory. Powyższa metoda prowadzi do powstania
materiałów węglowych z uporządkowaną strukturą mezoporowatą [57].
14
Polimer A
mieszanie
(„blending”)
karbonizacja
![]()
Polimer B
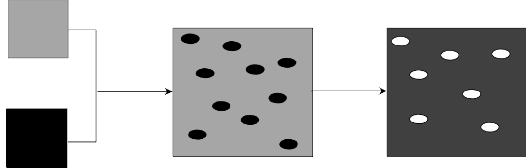
![]()
Materiał węglowy
Rysunek 3. Kontrola struktury porowatej materiałów polimerowych metodą
„blending” [58]
M. Inagaki i H. Konno zaproponowali innowacyjną metodę otrzymywania porowatych adsorbentów węglowych z szeregu surowców syntetycznych bez etapu aktywacji fizycznej. Mieszanina prekursora węgla (poli(chlorek winylu), poli(alkohol winylowy), poli(tereftalan etylenu), itd.) oraz materiału ceramicznego (SiO2, TiO2, MgO lub różne fazy Al2O3) została poddana jedynie wygrzewaniu w zakresie temperatur 600 –
1200 °C, w atmosferze gazu inertnego [59, 60, 61]. Struktura materiałów węglowych była
silnie zależna od rodzaju zastosowanego związku ceramicznego. Powyższą zależność, w szczególności można było zauważyć w przypadku węgli otrzymanych z mieszaniny zawierającej różne fazy Al2O3. [60]. W przypadku zastosowania w mieszaninie wyjściowej materiału ceramicznego w postaci MgO, uzyskany materiał węglowy po procesie karbonizacji został przemyty roztworem kwasu siarkowego, w celu oddzielenia stałej pozostałości nieorganicznej. Otrzymany materiał węglowy charakteryzował się silnie rozwiniętą powierzchnią właściwą, z dużym udziałem mikroporów. Zależność ta tłumaczona jest utrudnionym skurczem węgla formowanego z polimeru, który osadzony jest w postaci cienkiego filmu na powierzchni cząstek MgO, podczas procesu ogrzewania mieszaniny [61]. Znaczący wpływ na rozwój struktury porowatej otrzymanych materiałów węglowych, miał również udział wagowy tlenku magnezu w mieszaninie wyjściowej z PVA. Zaobserwowano, że wraz ze wzrostem udziału MgO w mieszanie wyjściowej miało miejsce wyraźne rozwinięcie struktury mikroporowatej materiałów węglowych [ 60].
Powyższą metodę rozszerzono o zastosowanie w mieszanie wyjściowej prekursorów
MgO (Tabela 1), ulegających rozkładowi termicznemu z wydzieleniem produktów
15
gazowych. Ponadto, zastosowanie takich prekursorów MgO jak cytrynian, octan czy glukonian magnezu (Mg3(C6H5O7)2, Mg(CH3COO)2, Mg(C11H22O14) umożliwiło zwiększenie wydajności węglowej, jako że rozkład wyżej wymienionych związków zachodził z wydzieleniem węgla. Węgle aktywne otrzymane powyższą metodą charakteryzują się dużą powierzchnią właściwą (do 2000 m2/g) oraz silnie rozwiniętym systemem porów. Dowiedziono, że rozmiar mezoporów utworzonych w materiale węglowym pokrywał się z wielkością krystalitów MgO. Poprzez odpowiedni dobór prekursora tlenku magnezu w mieszaninie wyjściowej można kontrolować rozmiar i zawartość mezoporów w otrzymanym materiale węglowym.
Tabela 1. Wpływ prekursorów tlenku magnezu na rozwój struktury porowatej materiałów węglowych
* PVP – poli(winylopirolidon), PAA – poli(akrylamid)
Rozwój struktury mikroporowatej w dużym stopniu zależał od budowy prekursora węgla oraz warunków procesu karbonizacji (szybkość grzania). Materiały poddane powolnemu procesowi ogrzewania (5 °C/min), nie charakteryzowały się tak znaczącym udziałem mikroporów w strukturze węgla, jak materiały ogrzewane z szybkością
10 °C/min. Materiały, w których prekursorem węgla był poli(alkohol winylowy) (PVA) odznaczały się lepiej rozwiniętą mikroporowatość, w porównaniu z materiałami otrzymanymi z mieszaniny wyjściowej zawierającej poli(tereftalan etylenu) (PET) lub hydroksypropylocelulozą (HPC). Jednakże, autorzy nie potrafili wyjaśnić, dlaczego
w przypadku zastosowania prekursora węgla w postaci dwóch pozostałych polimerów,
16
materiały charakteryzowały się większą zawartością mezoporów. Nie zaobserwowali również, wpływu gazowych produktów rozkładu zastosowanych prekursorów MgO na strukturę materiału węglowego. Wadą powyższej metody, mogłaby się wydawać konieczność wypłukiwania stałej pozostałości z materiału węglowego. Jednakże, w tym przypadku zostały zastosowane niekorozyjne roztwory kwasów. Ponadto, w przypadku użycia roztworu kwasu cytrynowego lub octowego, stwierdzono możliwość ponownego wykorzystania uzyskanego cytrynianu lub octanu magnezu (po procesie płukania materiału węglowego) w mieszaninie wyjściowej z prekursorem węgla (PVA) (Tabela 2).
Tabela 2. Ponowne wykorzystanie prekursora MgO [61]
![]()
Kwas octowy Kwas cytrynowy
![]()
![]()
![]()
![]()
Octan magnezu/PVA Cytrynian magnezu/PVA
Ilość
cykli
SBET
[m2/g]
Wydajność
węglowa
![]()
[%wag.]
SBET
[m2/g]
Wydajność
węglowa
[%wag.]
1 1210 9,8 1402 26,8
2 1185 9,6 1468 25,2
3 1262 9,3 1423 25,4
4 1203 10,1 1415 26,1
5 1249 9,1 1481 25,4
![]()
Dowiedziono, że MgO użyty w procesie, może być w 100% odzyskany. Powierzchnia właściwa i wydajność węglowa otrzymanych materiałów, po 5 cyklach pozostawała praktycznie bez zmian (Tabela 2).
Jak wspomniano w rozdziale 1.1 najczęściej stosowanymi surowcami w produkcji materiałów węglowych są różnorodne materiały pochodzenia organicznego, a rzadziej materiały syntetyczne. Powyższa metoda stwarza nowe możliwości odpadowym polimerom, w procesie otrzymywania materiałów węglowych o ściśle kontrolowanej strukturze porowatej i doborze parametrów adsorpcyjnych w selektywnym oczyszczaniu wody i powietrza.
1.4 Przetwarzanie odpadów z poli(tereftalan etylenu)
Zmniejszające się zasoby surowców naturalnych oraz masowy wzrost produkcji i zużycia tworzyw sztucznych powoduje, że te ostatnie, ze względu na znaczny udział
17
objętościowy w odpadach stają się materiałami uciążliwymi. Z drugiej strony, ze względu na zalety, takie jak termostabilność, przejrzystość, niskie koszty produkcji, ich konsumpcja stale rośnie [65].
Jako materiały syntetyczne do produkcji opakowań najczęściej stosuje się: poli(chlorek winylu) (PCV), poli(tereflatan etylenu) (PET), polipropylen (PP), polietylen (PE), polistyren (PS). Poli(tereftalan etylenu) (PET) od kilkudziesięciu lat stosuje się do produkcji folii i włókien poliestrowych [66]. Tworzywo to stało się również najpopularniejszym surowcem w produkcji opakowań do napojów. W skali przemysłowej, PET otrzymuje się najczęściej metodą wymiany estrowej między estrem
metylowym kwasu tereftalowego, a glikolem etylenowym. Proces ten przebiega w dwóch etapach, zakończonych polikondensacją tereftalanu [67]. Masowa produkcja opakowań wykonanych z PET przekroczyła możliwości ich selektywnej zbiórki. Utylizacja powyższego odpadu, biorąc pod uwagę jego niską bio- i fotobiodegradowalność, stanowią poważny problem dla krajów uprzemysłowionych [65]. Z ekonomicznego punktu widzenia najkorzystniejszym sposobem utylizacji odpadów PET jest oczyszczenie i ponowne przetwórstwo otrzymanego recyklatu, w celu uzyskania poszukiwanych wyrobów. Do tego celu są jednak potrzebne odpady o dużej czystości. Recykling PET ze użytych butelek jest utrudniony, gdyż wymaga usunięcia domieszek (resztki napojów, etykiety papierowe, klej akrylowy do etykiet, uszczelki z folii aluminiowej, itp.), a także elementów z innych tworzyw sztucznych [66]. W ostatnich latach nasiliły się badania nad otrzymywaniem i charakterystyką węgli aktywnych otrzymanych z różnych polimerów, m.in. z PET [68, 69, 70, 71, 72]. Materiał ten bardzo trudno ulega naturalnemu rozkładowi,
a jednocześnie charakteryzuje się wysoką zawartością węgla (ponad 60 %wag.) [73].
Jednakże, istotnym faktem z punktu widzenia otrzymywania adsorbentów węglowych jest niewielka wydajność węglowa tego surowca, osiągająca jedynie około 20%, a stały produkt pirolizy wykazuje powierzchnię właściwą jedynie rzędu od kilkunastu do ok.
250 m2/g [74]. Należy podkreślić, że uzyskanie materiału o wysokiej powierzchni
właściwej wymaga zastosowanie dodatkowego etapu, jakim jest proces aktywacji, któremu nieodłącznie towarzyszy dodatkowy, często bardzo znaczny ubytek masy. Taka droga otrzymywania jest, zatem obarczona bardzo niską całkowitą wydajnością produktu, wynoszącą kilka procent [75], co w porównaniu z węglami aktywnymi otrzymywanymi z surowców konwencjonalnych jest bardzo niekorzystne i znacznie
wpływa na obniżenie opłacalności produkcji. Ze względu na bardzo niską wydajność
18
produktu, prowadzi się badania w kierunku opracowania alternatywnych sposobów otrzymywania adsorbentów węglowych z PET. Badania te mają na uwadze, zarówno warunki prowadzenia procesu (temperatura i czas karbonizacji), rodzaj stosowanego aktywatora oraz właściwości otrzymanego węgla (powierzchnia właściwa, rozmiar porów).
Najczęściej proponowanym sposobem wytwarzania węgla aktywnego z PET jest dwustopniowy proces obejmujący pirolizę rozdrobionego prekursora w strumieniu gazu inertnego, a następnie aktywację w strumieniu dwutlenku węgla lub pary wodnej [65, 76,
77]. Z badań nad wpływem warunków procesu na rozwój struktury porowatej
materiałów węglowych otrzymanych z PET, aktywowanego CO2, wynika, że zarówno temperatura jak i czas trwania aktywacji ma znamienny wpływ na rozwój porowatości otrzymanych materiałów węglowych. Stosunkowo niewielki wzrost czasu aktywacji (z 4 do 5 godzin) w temperaturze 940 °C, zwiększa znacząco powierzchnię właściwą i objętość mikroporów [76]. Ponadto, zwiększenie stopnia wypału prowadzi do wyraźnego wzrostu powierzchni właściwej (ponad 2400 m2/g) oraz udziału mikroporów i mezoporów w otrzymanych materiałach węglowych [78].
Do otrzymywania węgla aktywnego z PET zastosowano również aktywację chemiczną z użyciem H2SO4 [79], Ca(NO3)2∙4H2O [77], z późniejszym użyciem gazowego aktywatora. Badania nad procesami aktywacji chemicznej miały na celu zmniejszenie strat masy prekursora (wzrost wydajności węglowej) oraz rozwinięcie struktury mezoporowatej materiałów węglowych. Jednakże stosowane procedury były niekiedy dość złożone. H. Tamon i in. [77] zbadali wpływ czasu oraz rodzaju aktywacji na powierzchnię i porowatość węgla aktywnego otrzymanego z PET. Porównano dwie metody otrzymywania węgla aktywnego: dwustopniowy proces obejmujący pirolizę rozdrobionego PET w strumieniu gazu inertnego, a następnie aktywację w strumieniu pary wodnej oraz proces obejmujący aktywacje chemiczną związkiem wapnia, i końcową aktywację parą wodną, poprzedzoną kwaśnym myciem (HCl) otrzymanego produktu. Materiały poddane wstępnej obróbce charakteryzowały się znacznie większą zawartością mezoporów, podobnie jak w przypadku zastosowania H2SO4, jako czynnika aktywującego.
W celu zmniejszenia ubytku masy karbonizatu otrzymanego z PET, prekursor węgla poddano modyfikacji FeCl3 [80, 81]. Obecność chlorku żelaza (III) znacząco zwiększyła wydajność, która wyniosła blisko 50% (w porównaniu z 20% wydajnością w przypadku
PET niemodyfikowanego). Zastosowanie roztworu FeCl3 wiąże się z obecnością
19
związanego chloru w karbonizacie. Modyfikację PET przeprowadzono także za pomocą innych soli – szczawianu i siarczanu żelaza. PET występnie gotowany w roztworze szczawianu żelaza wykazał większą podatność na zwęglanie w porównaniu z materiałem obrabianym w FeCl3 czy FeSO4. Ogólnie, wprowadzenie do polimeru soli żelaza wpłynęło korzystnie na przebieg procesu zwęglania, czyli spowodowało zwiększenie wydajności procesu. Zastosowanie szczawianu żelaza (III) pozwala na eliminację z gazów odlotowych HCl i tlenku siarki pojawiającej się w przypadku użycia FeCl3 i FeSO4.
Ciekawą propozycją przetwarzania polimeru na węgiel aktywny, jest depolimeryzacja
PET poprzez działanie wodorotlenkami metali alkalicznych (KOH lub NaOH) i następnie karbonizacja stałej pozostałości złożonej z tereftalanu metalu alkalicznego z domieszką odpowiedniego wodorotlenku [82, 83]. Proces termiczny prowadzony w temperaturze 650 °C lub 800 °C w strumieniu azotu, prowadzi do uzyskania węgli aktywnych. W przypadku zastosowania KOH i wyższej z tych temperatur, węgle wykazywały powierzchnie właściwe powyżej 1000 m2/g, czyli odpowiadające handlowym adsorbentom węglowym. Wspomniany proces umożliwia połączenie procesu karbonizacji i aktywacji w jeden etap, co skutkuje wzrostem wydajności węglowej i hamuje wydzielanie się lotnych związków. W porównaniu z metodą karbonizacji PET i aktywacją CO2 jest to zaletą. Materiały otrzymane tą metodą wykazują wysoką pojemność adsorpcyjną oraz stosunkowo jednorodne i wąskie mikropory.
W ostatnich latach wiele uwagi poświęca się zastosowaniu różnych związków magnezu, do otrzymywania porowatych materiałów węglowych z polimerów. Szczegółowo opisana metoda (rozdział 1.3), z zastosowaniem mieszaniny syntetycznego prekursora węgla z związkami magnezu, pozwala otrzymać w jednoetapowym procesie materiały węglowe o dużej powierzchni właściwej (1000 –
2000 m2/g), z dobrze rozwiniętym systemem porów [61]. Oprócz zbadania wpływu
budowy użytego związku magnezu oraz jego udziału wagowego w mieszaninie wyjściowej na rozwój struktury porowatej materiałów węglowych, autorzy przeprowadzili badania nad wpływem sposobu przygotowania materiału wyjściowego na strukturę materiału węglowego [62]. Mieszaninę wyjściową przygotowano w dwojaki sposób, tj. w postaci proszku lub roztworu, z którego następnie odparowano rozpuszczalnik. Otrzymany materiał karbonizowano w 900 °C, w atmosferze gazu
obojętnego. Następnie otrzymany karbonizat poddano kwaśnemu myciu (kwas
20
siarkowy), w celu odmycia stałej pozostałości w postaci tlenku magnezu. Sposób przygotowania mieszaniny wyjściowej wpłynął znacząco na porowatość otrzymanych węgli aktywnych. Przy zastosowaniu materiału wyjściowego w postaci proszkowej, uzyskano materiały o charakterze mikroporowatym. Natomiast, w przypadku mieszaniny wyjściowej w postaci roztworu uzyskane węgle miały strukturę porowatą o większym udziale mezoporów. Na podstawie dokonanego przeglądu literatury można stwierdzić, że wartym rozwinięcia jest opracowanie jednoetapowej metody otrzymywania materiałów węglowych z polimerów, łączącego w sobie proces karbonizacji i aktywacji fizycznej. Przetwarzanie PET w węgle aktywne daje podwójną korzyść z punktu widzenia ochrony środowiska: może być sposobem usunięcia uciążliwego odpadu, a uzyskany produkt może znaleźć zastosowanie w procesach oczyszczania wody i powietrza. Jak wynika z szeregu przedstawionych powyżej publikacji, poprzez odpowiedni dobór warunków procesu oraz rodzaju materiału ceramicznego, można wpływać na strukturę porowatą otrzymanych materiałów węglowych. Kontrolowana zawartość mikro- i mezoporów w strukturze materiału węglowego umożliwia selektywną adsorpcję zanieczyszczeń, zarówno z fazy ciekłej jak i gazowej.
1.5 Węgle aktywne w selektywnej adsorpcji zanieczyszczeń wody
Szybki rozwój przemysłu prowadzi do wzrostu poziomu zanieczyszczenia wód. Ścieki pochodzące z aglomeracji miejskich, zwiększające się zużycie środków powierzchniowo czynnych, nawozów sztucznych, rozkład materii organicznej w dużej mierze przyczyniają się do wprowadzenia znacznych ilości różnorodnych zanieczyszczeń do wód powierzchniowych. W procesach oczyszczania ścieków stosuje się szereg fizycznych, biologicznych i chemicznych metod, jak np. filtracja, koagulacja, utlenianie, wymiana jonowa, procesy membranowe czy adsorpcja [84]. Proces adsorpcji z wykorzystaniem węgli aktywnych stanowi zazwyczaj jeden z ostatnich etapów cyklu oczyszczania, gdzie różnorodność zanieczyszczeń obecnych w wodzie, w niskich stężeniach, powoduje, że inne metody mogą nie przynieść oczekiwanych efektów.
Adsorpcja na węglu aktywnym jest technologią o szerokim spektrum zastosowań, tj. w procesach usuwania przede wszystkim organicznych, ale także nieorganicznych zanieczyszczeń wody [14]. Zdolności adsorpcyjne materiałów węglowych w procesach
usuwania zanieczyszczeń z fazy ciekłej, determinują takie czynniki jak:
21
– struktura porowata adsorbentu, w tym rozkład objętości i rozmiaru porów oraz
powierzchnia właściwa [85];
– natura fizykochemiczna powierzchni materiału (obecność grup funkcyjnych [86],
oddziaływania elektrostatyczne [87], hydrofobowość [88], zawartość popiołu [85]);
– masa cząsteczkowa, budowa, właściwości chemiczne adsorbatu [89];
– parametry uzdatnianej wody (pH, stężenia jonów w roztworze [90], zawartość tlenu
[91]).
Zastosowanie węgli aktywnych w procesach oczyszczania wody jest tematem licznych badań [92, 93, 94]. Przykładem, może być usuwanie jonów metali ze ścieków i wody pitnej. M. O. Corapcioglu i C. P. Huang [95] stwierdzili, że na efektywność sorpcji względem takich jonów metali jak Cu (II), Pb (II), Ni (II) i Zn (II) ma wpływ rodzaj zastosowanego węgla, pH roztworu oraz ładunek powierzchniowy węgla. V. C. Srivastava i in. [96] określili maksimum sorpcji jonów metali Cd (II), Ni (II) i Zn (II) przy wartości pH 6, z wykorzystaniem komercyjnego węgla aktywnego, otrzymanego z orzecha kokosowego. Ponadto, obecność tlenowych grup funkcyjnych powoduje, że powierzchnia węgla staje się polarna, co sprzyja wymianie jonowej i przyczynia się do efektywnej sorpcji różnych jonów metali [14]. M. Kobya i in. [97] zbadali usuwanie jonów metali w zakresie pH 1 – 6, przez adsorpcję na węglach
aktywnych otrzymanych z pestek moreli, aktywowanych kwasem siarkowym,. Uzyskano ponad 99% skuteczność usuwania jonów Cr (VI), Cd (II) oraz Pb (II). W przypadku adsorpcji jonów Cr (VI) badacze zauważyli wyraźny wzrost adsorpcji w miarę obniżania pH. Przypisali to redukcji Cr (VI) do Cr (III) [98]. Usuwanie jonów Ni2+, Co2+, Cd2+, Pb2+ oraz Cu2+ było maksymalne przy pH 3 – 6. Natomiast przy niższych wartościach pH zdolność adsorpcyjna materiałów była wyraźnie niższe.
Rezultaty te zostały wyjaśnione w oparciu o zmiany ładunku powierzchni węgla, ze zmianą pH roztworu. Przy niskim pH powierzchnia węgla miała ładunek dodatni i tym samym zachodziły elektrostatyczne oddziaływania odpychania między kationami i dodatnio naładowaną powierzchnią [99].
Obecność różnych związków organicznych w wodzie (fenol, pestycydy,
węglowodory aromatyczne czy barwniki), niesie za sobą poważne zagrożenie dla środowiska naturalnego. Fenol i jego pochodne stanowią grupę zanieczyszczeń, które są obecne w ściekach pochodzących z przemysłu chemicznego, petrochemicznego, farmaceutycznego, produkcji farb, barwników czy tworzyw sztucznych [100].
Mechanizmy adsorpcji fenoli na węglach aktywnych były i są tematem obszernych
22
badań [101, 102, 103]. Stwierdzono, że zdolność adsorpcyjna węgla względem fenolu, silnie zależy od pola powierzchni adsorbentu i jego struktury mikroporowatej [103]. Adsorpcja fenolu najbardziej skutecznie zachodzi w mikroporach, o wymiarach zbliżonych do wielkości adsorbowanej cząsteczki. Powodem tego są najsilniejsze oddziaływania między adsorbatem a powierzchnią ścianek porów [104]. Istotny wpływ na adsorpcję związków fenoli ma również pH roztworu. Jak wynika z badań przytoczonych przez innych autorów [105, 106], adsorpcja fenolu zachodzi najefektywniej w środowisku kwaśnym. W środowisku zasadowym fenole ulegają dysocjacji, tworząc ujemnie naładowane jony fenolanowe, natomiast tlenowe grupy funkcyjne zawarte na
powierzchni adsorbentu są częściowo zjonizowane, nadając powierzchni ujemny ładunek. Powoduje to elektrostatyczne odpychanie między powierzchnią węgla aktywnego, a jonami fenolowymi, co w konsekwencji zmniejsza adsorpcję fenoli [14]. Z badań nad wpływem powierzchniowych grup tlenowych na adsorpcję fenolu z roztworów wodnych, z użyciem węgli aktywnych utlenionych nadsiarczanem amonu [107]. Utlenienie węgli powoduje zmniejszenie adsorpcji fenolu. Ma to związek, z obecnością na powierzchni materiału kwasowych grup powierzchniowych. Podobne obserwacje opisał R. C. Bansal i in. [108], badając adsorpcja na węglach aktywnych utlenionych kwasem azotowym (V), nadtlenodisiarczanem (VI) amonu i nadtlenkiem wodoru. Maksymalne obniżenie adsorpcji fenolu obserwowano w przypadku utlenienia węgla kwasem azotowym (V), który tworzył na powierzchni adsorbentu największe ilości kwasowych grup powierzchniowych. W celu określenia rodzaju powierzchniowych grup tlenowych, mających wpływ na adsorpcję fenolu, materiały węglowe poddawano odgazowaniu w narastającej temperaturze od 400 °C do 950 °C. Adsorpcja fenolu wzrastała w miarę wzrostu temperatury odgazowania i osiągnęła maksimum dla węgli ogrzewanych w 650 °C. W trakcie ogrzewania materiału węglowego następuje rozkład grup tlenowych, z wydzieleniem CO2 (w temperaturze
350 °C – 750 °C) lub CO (w temperaturze 500 °C – 900 °C). Grupy rozkładające się
z wydzieleniem CO2 (karboksylowe lub laktonowe), nadawały powierzchni węgla polarny i hydrofilowy charakter, a tym samym przyczyniały się do hamowania adsorpcji fenolu. Powierzchniowe grupy tlenowe, rozkładające się z wydzieleniem CO (chinonowe) zwiększały powinowactwo powierzchni węgla do fenolu, poprzez nadanie charakteru hydrofobowego. J. L. Figueiredo i in. [109] zbadali zdolność adsorpcyjną aktywnych włókien węglowych (ACF, ang. Activated Carbon Fiber) względem fenolu.
Materiały te były otrzymane przez aktywację CO2 lub H2O. Zauważono silną zależność
23
pomiędzy strukturą mikroporowatą materiałów, a ilością zaadsorbowanego fenolu. Ponadto, adsorpcja fenolu była znacząco wyższa na materiałach aktywowanych CO2. Zaobserwowano, że adsorpcja obejmowała zarówno sorpcję fizyczną jak i chemiczną. Zbadano zależność pomiędzy zawartością tlenowych grup funkcyjnych, a ilością fizycznie i chemicznie adsorbowanego fenolu na powierzchni materiału węglowego. Stwierdzono, że ze zwiększeniem ilości tlenowych grup funkcyjnych na powierzchni ACF, chemisorpcja fenolu malała. Zauważono, że chemisorpcja fenolu jest ograniczona do miejsc wolnych od tlenu, zlokalizowanych przede wszystkim na krawędziach warstw grafenowych, a fizysorpcja zachodzi na całej powierzchni adsorbentu.
Jednym z głównych problemów w procesie oczyszczania ścieków oprócz wyżej omawianego zanieczyszczenia są związki powszechnie wykorzystywane w przemyśle włókienniczym, farbiarskim, jak i papierniczym. Barwniki obecne w ściekach mogą wykazywać działanie mutagenne i kancerogenne, gdyż w warunkach redukcyjnych mogą uwalniać się szkodliwe aminy aromatyczne [110]. Ścieki barwne zakłócają równowagę biologiczną wód, hamują procesy rozkładu biochemicznego poprzez wiązanie tlenu rozpuszczonego w wodzie. Ponadto, w znacznym stopniu ograniczają przenikanie światła, wpływając na osłabienie procesów asymilacji roślin [14]. Adsorpcja barwników na węglu aktywnym została zbadana pod wieloma aspektami. Badano m.in. wpływ charakteru powierzchni węgli (struktury porowatej oraz polarności), pH roztworu czy rodzaju zastosowanego barwnika (kationowy, anionowy) [111]. Stwierdzono istotny wpływ wartości pH roztworu w procesie adsorpcji barwników. J .J. M. Órfão i in. [112] badający adsorpcję czerwieni reaktywnej 241 na węglach aktywnych, wyjaśnili zależność ładunku powierzchni węgla od pH roztworu. Stwierdzono, że to potencjał zeta powierzchni węgla określa jego zdolność adsorpcyjną. Wykazano dla wartości pH roztworu barwika wyższych od pH, dla którego gęstość ładunku elektrycznego na powierzchni jest równa zero (pH>pHPZC), elektrostatyczne odpychanie pomiędzy anionowym barwnikiem i ujemnie naładowaną powierzchnią węgla zmniejsza adsorpcję barwnika. W przypadku barwników kationowych jak np. błękitu metylenowego [113], w miarę podwyższania pH roztworu powierzchnia węgla przyjmuje ładunek ujemny, co w rezultacie prowadzi do większej adsorpcji kationowych cząsteczek barwnika.
Niemniej ważną rolę w adsorpcji zanieczyszczeń z roztworów wodnych odgrywa
rozkład i wielkość porów adsorbentów węglowych. H. Tamai i in. [114] zbadali zdolność
adsorpcyjną mezoporowatych (Y-ACF) i mikroporowatych (A-20) włókien węglowych
24
względem kwasowych, obojętnych oraz zasadowych barwników. Barwniki o małych rozmiarach cząsteczki były adsorbowane w znacznych ilościach na obu rodzajach materiałów. W porównaniu z materiałem mikroporowatym, adsorpcja barwników zasadowych, o stosunkowo dużych rozmiarach cząsteczek, była wyraźnie większa na mezoporowatym Y-ACF. Ponadto, zdolność adsorpcyjna węgla aktywnego względem barwników zasadowych malała wraz ze wzrostem pH, w miarę jak rozmiary cząsteczek barwnika rosły i układały wg. następującego porządku Direct Yellow 50 (2,39 nm) > Direct Black 19 (30,4 nm) > Direct Yellow 11 (31,2 nm). Wysoką zdolność adsorpcyjną węgla względem barwników zasadowych przypisano przyciąganiu elektrostatycznemu między powierzchnią materiału, a cząsteczkami barwnika oraz dyfuzją molekuł barwnika do struktury porowatej materiału. E. Lorence – Grabowska
i G. Gryglewicz [115] potwierdziły, że struktura materiału węglowego jest kluczowym
czynnikiem odgrywającym rolę w adsorpcji barwników. Zaobserwowano, że adsorpcja barwnika o dużych cząsteczkach jak czerwieni congo rosła wraz ze wzrostem objętości mezoporów. Również adsorpcja naturalnych związków organicznych jak kwasy humusowe [116] zależy silnie od struktury porowatej węgla. Wykazano, że węgle z dobrze rozwiniętym system mezoporów charakteryzują się wysoką zdolnością adsorpcyjną względem naturalnej materii organicznej [117], charakteryzującej się obecnością struktur o dużych masach cząsteczkowych.
Przytoczone powyżej informacje wskazują, że mechanizm adsorpcji związków organicznych z roztworów wodnych na materiałach węglowych był różnie interpretowany. Różnicom w ilości zaadsorbowanych zanieczyszczeń zostały przypisane różne czynniki takie jak wpływ pH, obecność powierzchniowych grup funkcyjnych czy oddziaływania elektrostatyczne. Niemniej istotny wpływ na proces adsorpcji miał charakter adsorbentu, a w szczególności struktura porowata (rozkład objętości porów wg. ich szerokości, całkowita objętość porów). W doborze węgla aktywnego do specyficznego procesu adsorpcji należy także zwrócić uwagę na rozmiar cząsteczek adsorbatu. W przypadku zastosowania materiału węglowego o dominującym charakterze mikroporowatym część porów może być niedostępna dla cząsteczek adsorbatu, o dużych rozmiarach. Dlatego ważne jest opracowanie metody otrzymywania adsorbentów z możliwością łatwej kontroli zawartości mikroporów i mezoporów, co z pewnością będzie miało znaczący wpływ na selektywną adsorpcję
zanieczyszczeń o szerokim zakresie mas cząsteczkowych.
25
Część doświadczalna
![]()
2. Cel i zakres pracy
Celem pracy było otrzymanie porowatych materiałów węglowych w jednoetapowym procesie, polegającym na karbonizacji prekursora węgla w postaci poli(tereftalanu etylenu) (PET) z dodatkiem związku nieorganicznego, z pominięciem klasycznego etapu aktywacji fizycznej. W tym celu należało użyć związku nieorganicznego, ulegającego rozkładowi termicznemu z wydzieleniem produktów gazowych będących typowymi czynnikami aktywującymi stosowanymi w procesie aktywacji fizycznej (CO2, para wodna), a przy tym powodującego tworzenie się struktury porowatej węgla.
Odpowiedni dobór jakościowy i ilościowy reagentów może być sposobem wpływania na strukturę porowatą otrzymywanych adsorbentów. Jest to bardzo ważne z punktu widzenia otrzymywania adsorbentu o określonych, powtarzalnych właściwościach. Oprócz tego, z racji narastających problemów związanych z zanieczyszczeniem środowiska naturalnego, otrzymanie materiałów służących ochronie środowiska z surowców będących zanieczyszczeniem jak np. odpadowy PET, jest bardzo ważnym aspektem. Opracowanie metody, która pozwalałaby na kontrolę zawartości mikroporów i mezoporów w węglach aktywnych jest ważna ze względu na wykorzystanie adsorbentów do selektywnej adsorpcji zanieczyszczeń o szerokim zakresie mas cząsteczkowych. Dlatego też dodatkowym celem było wyjaśnienie wpływu produktów rozkładu użytych związków magnezu (stałego – tlenek magnezu i gazowych – CO2/H2O) na rozwój porowatości oraz mechanizmu tworzenia porów w badanych materiałach węglowych. Kolejnym celem pracy badawczej było badanie wpływu wybranych związków magnezu stosowanych w mieszaninie z PET na wydajność
tworzenia węgla aktywnego.
27
3. Charakterystyka stosowanych materiałów
3.1 Komercyjny węgiel aktywny CWZ-35
Materiałem porównawczym był handlowy pylisty węgiel aktywny o symbolu CWZ-35, otrzymany metodą parowo-gazowej aktywacji węgla drzewnego, krajowej firmy Gryfskand Sp. z o.o..
3.2 Prekursor węgla - poli(tereftalan etylenu) (PET)
Surowcem wyjściowym w procesie otrzymywania materiałów węglowych był granulat poli(tereftalanu etylenu), zakupiony od firmy Elana S.A., Toruń.
3.3 Związki magnezu
W Tabeli 3 zestawiono związki magnezu użyte w mieszanie wyjściowej
z poli(tereftalanem etylenu).
3.4 Modelowe zanieczyszczenia organiczne
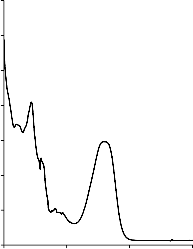
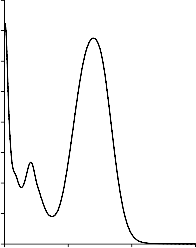
W Tabeli 4 zestawiono modelowe zanieczyszczenia organiczne, wykorzystane, jako adsorbaty, podczas badania właściwości sorpcyjnych otrzymanych materiałów węglowych. Na Rysunku 4 przedstawiono widma absorpcyjne UV-Vis zastosowanych adsorbatów. Posłużyły one do wyznaczenia analitycznych długości fali, przy których prowadzono pomiary spektrofotometryczne.
28
a) 1,6
1,4
1,2
1,0
0,8
0,6
0,4
0,2
0,0

200 250 300 350 400
Długość fali, nm
c) 0,35
b) 4,0
3,5
3,0
2,5
2,0
1,5
1,0
0,5
0,0
200 400 600 800
Długość fali, nm
0,30
0,25
0,20
0,15
0,10
0,05
0,00
200 400 600 800
Długość fali, nm
Rysunek 4. Widmo absorpcyjne UV-Vis: a) fenolu, b) czerwieni anilinowej (BR 18), c) czerwieni reaktywnej (RR 198)
29![]()
Tabela 3. Zastosowane związki magnezu
![]()
Związek magnezu | Producent | Wzór sumaryczny | Numer CAS | Masa molowa [g/mol] |
Lekki zasadowy węglan magnezu (BMC) (cz.d.a.) | POCH S.A. | 3MgCO3·Mg(OH)2·3H2O | 12125-28-9 | 365,24 |
Wodorotlenek magnezu (cz.d.a.) | Fluka | Mg(OH)2 | 1309-42-8 | 58,32 |
Tlenek magnezu (cz.d.a.) | Chempur | MgO | 1309-48-4 | 40,30 |
![]()
![]()
Tabela 4. Modelowe zanieczyszczenia organiczne
![]()
Adsorbat | Producent | Wzór sumaryczny | Numer CAS | Masa molowa [g/mol] | λmax (nm) |
Czerwień reaktywna 198 (RR198) | Boruta-Kolor Sp. z o. o. | C27H18ClN7Na4O15S5 | 145017-98-7 | 968,21 | 516 |
Czerwień anilinowa 18 (BR18) | Boruta-Kolor Sp. z o.o. | C19H25ClN5O2 | 14097-03-1 | 390,89 | 480 |
Fenol | Sigma-Aldrich | C6H6O | 8002-07-1 | 94,11 | 270 |
![]()
30
4. Preparatyka węgli aktywnych
4.1. Przygotowanie mieszaniny związku nieorganicznego z PET
Porcję granulatu PET umieszczono w komorze mielącej młynka analitycznego A11
Basic, firmy IKA w ERKE GmbH. Następnie do komory wlano ok. 50 cm3 ciekłego azotu. Po odparowaniu czynnika chłodzącego, granulat zmielono mechanicznie do postaci proszku. Rozdrobniony PET zmieszano z wybranymi związkami magnezu w trzech proporcjach masowych (Tabela 5).![]()
Tabela 5. Jakościowy i ilościowy skład mieszanin wyjściowych stosowanych w badaniach
![]()
Związek magnezu/PET | Udział wagowy [%wag.] |
3MgCO3·Mg(OH)2·3H2O/PET | 30/70 50/50 70/30 |
Mg(OH)2/PET | 30/70 50/50 70/30 |
MgO/PET | 30/70 50/50 70/30 |
![]()
W celu uzyskania homogenicznego składu, każdą z mieszanin ogrzano do temperatury
265 °C (temperatura topnienia PET) [118] z prędkością grzania 5 °C/min w piecu rurowym Naberthern R40/250/12-C40 w atmosferze gazu inertnego (argon, czystość
99,999%, 25 cm3/min), a następnie utrzymywano w tej temperaturze przez 1 godzinę.
Otrzymaną mieszaninę schłodzono i ponownie zmielono. Powyższą procedurę powtórzono dwukrotnie.
4.2. Karbonizacja mieszanin związek magnezu/PET
Karbonizację surowej mieszaniny (związek magnezu/PET) otrzymanej według procedury 4.1, przeprowadzono w piecu rurowym Naberthern R40/250/12-C40 w atmosferze gazu inertnego (Ar, 25cm3/min). Naważkę każdej z mieszanin (ok. 6 g)
31
umieszczano w grafitowej łódce, w prowadzano do centralnej części rury kwarcowej
pieca (strefa stałej temperatury) i ogrzewano do temperatury 550 °C lub 700 °C lub
850 °C (10 °C/min). Próbkę utrzymywano w docelowej temperaturze przez 1 godzinę. Następnie układ chłodzono do temperatury pokojowej.
4.3 Wydzielanie materiałów węglowych z otrzymanych karbonizatów
W celu uzyskania materiału węglowego niezawierającego pozostałości nieorganicznej, powstającej w trakcie procesu karbonizacji, otrzymany materiał hybrydowy przemywano roztworem mocnego kwasu. Porcje uzyskanych karbonizatów zalewano roztworem 10% kwasu solnego (0,1 dm3). Uzyskaną zawiesinę poddano ciągłemu mieszaniu przez 24 godziny. Następnie powstałą zawiesinę przemyto kilkakrotnie wodą destylowaną, aż do uzyskania stałego odczynu (pH ~6). W końcowym etapie zawiesinę poddano filtracji pod zmniejszonym ciśnieniem, w celu wyodrębnienia materiału węglowego, który następnie wysuszono w temperaturze 110 °C w atmosferze powietrza do stałej masy (12 godzin).
Procedurę otrzymywania docelowych materiałów węglowych przestawiono na
Rysunku 5.
32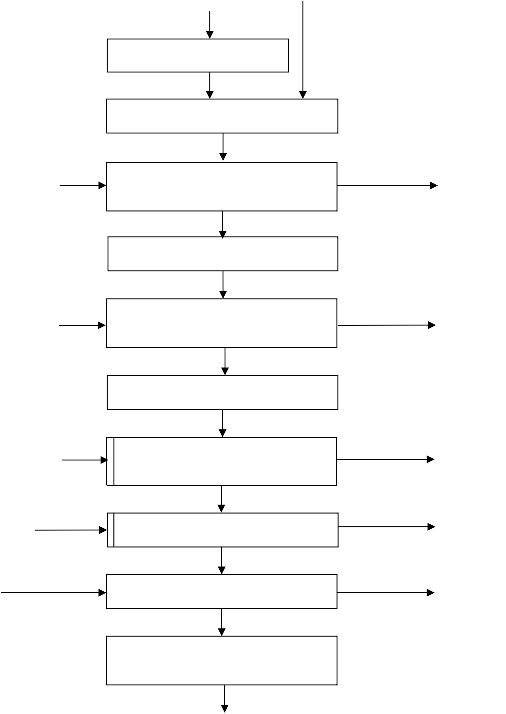
PET
związek magnezu
ROZDRABNIENIE
MIESZANIE
Ar OGRZEWANIE
(265 °C)
gazy odlotowe
MIELENIE
Ar OGRZEWANIE
(265 °C)
gazy odlotowe
MIELENIE
Ar PIROLIZA (550-850 °C)
gazy odlotowe
10% HCl
KWAŚNE MYCIE
roztwór MgCl
2
woda destylowana
MYCIE
roztwór MgCl2
SUSZENIE
(110 °C)
PRODUKT
Rysunek 5. Schemat otrzymywania materiału węglowego
33
5. Metody analityczne
5.1 Analiza termograwimetryczna (TG)
Wyznaczenie krzywych termograwimetrycznych PET, stosowanych związków nieorganicznych oraz ich mieszanin, przeprowadzono przy pomocy termograwimetru STA 449 C TGA firmy Netzsch, Niemcy, w atmosferze argonu (25 cm3/min). Badania termograwimetryczne polegały na rejestracji zmian masy podczas ogrzewania próbki (ok. 10 mg) do temperatury 850 °C (10 °C/min). Próbkę utrzymywano w końcowej temperaturze przez 1 godzinę. W celu określenia ilości pozostałości nieorganicznej (MgO) w otrzymanym materiale węglowym po procesie pirolizy, przeprowadzono dodatkowo analizę termograwimetryczną w atmosferze powietrza (25 cm3/min), przy wykorzystaniu analogicznego programu temperaturowego. Na podstawie różnicy mas otrzymanej po procesie analizy termograwimetrycznej w atmosferze argonu i powietrza oszacowano zawartość węgla w otrzymanych materiałach.
5.2 Dyfrakcja promieniowania rentgenowskiego (XRD)
Skład fazowy otrzymanych materiałów węglowych określono za pomocą dyfraktometru rentgenowskiego X’Pert PRO (Philips, Holandia) z użyciem promieniowania Cu Kα (λ=1,54056 Å). W oparciu o uzyskane dyfraktogramy wyznaczono także średnią wielkość krystalitów MgO zgodnie z równaniem Scherrera (Równanie 1).
Kλ![]()
D =
βcosθ
(1)
gdzie: D – średnia wielkość krystalitów MgO [nm];
K – stała Scherrera (K = 0,9);
λ – długość fali padającego promieniowania [Å];
θ – połowa kąta ugięcia [rad];
β – szerokość refleksu w połowie jego wysokości [°].
Uzyskane wyniki uwzględniały wyznaczoną doświadczalnie poprawkę aparaturową.
34
5.3 Pomiar izoterm adsorpcji/desorpcji azotu w 77 K
Parametry struktury porowatej otrzymanych materiałów węglowych określono na podstawie pomiarów izoterm adsorpcji/desorpcji azotu w 77 K na analizatorze Quadrasorb SI (Quantachrome, USA) w przedziale ciśnień względnych od ok. 10-3 do
0,99, wykorzystując azot o czystości 99,998%. Przed pomiarem izoterm, próbki odgazowano pod próżnią w temperaturze 290 °C w ciągu 24 godziny, pod obniżonym ciśnieniem 0 Torr. Z uzyskanych izoterm adsorpcji/desorpcji azotu wyznaczono następujące parametry struktury porowatej węgli aktywnych:
- SBET – powierzchnia właściwa obliczona z równania Braunauera-Emmeta-Tellera (BET) [119], wykorzystując część adsorpcyjną izoterm z przedziału ciśnień względnych od 0,05 do 0,50;
- wartości całkowitej powierzchni właściwej (Stotal) oraz powierzchni mikroporów (Smicro) i mezoporów (Sext) obliczono stosując metodę αs [120] gdzie αs jest standardową adsorpcją względną, zdefiniowaną jako stosunek wartości adsorpcji przy danym ciśnieniu względnym do wartości adsorpcji przy ciśnieniu względnym równym 0,4 dla
adsorbentu odniesienia;
- VmicroDR – objętość mikroporów obliczona według równania Dubinnina- Raduszkiewicza [121];
- Vtot 0,95 – całkowita objętość porów wyznaczona bezpośrednio na podstawie izotermy adsorpcji przy ciśnieniu względnym p/p0 = 0,95;
- objętość mezoporów – jako różnicę Vtot 0,95 i VmicroDR;
- funkcję rozkładu objętości porów obliczono wykorzystując metodę Barretta-Joynera- Halendy (BJH) w oparciu o równanie Kelvina [122].
5.4 Termoprogramowalna desorpcja (TPD)
Pomiar TPD został przeprowadzony za pomocą spektrometru desorpcji termicznej (TDS 1200, ESCO Ltd., Japonia) wyposażonego w spektrometr masowy. Próbkę poddano ogrzewaniu od temperatury pokojowej do 1000 °C (60 °C/min) pod ciśnieniem
35
~10-7 Pa. Pomiar TPD został przeprowadzony dla materiałów węglowych otrzymanych z mieszanin wyjściowych uprzednio ogrzanych do temperatury 700 °C, w atmosferze gazu obojętnego. Podczas pomiaru TPD monitorowano wydzielanie się czterech gazów z badanej próbki: H2, H2O, CO i CO2.
5.5 Rentgenowska spektroskopia fotoelektronów (XPS)
Skład jakościowy oraz ilościowy powierzchni badanych materiałów badany był za pomocą spektroskopii fotoelektronów (X-ray Photoelectron Spectroscopy – XPS). Badania zostały wykonane w systemie próżniowym firmy Prevac. W całym systemie poziom próżni utrzymywany był na poziomie 10-10 bar. Komora analizy wyposażona jest w hemisferyczny spektrometr elektronowy firmy Scienta, model SES 2002, pozwalający na analizę widma elektronów w zakresie energii 0,1 eV – 1500 eV, z rozdzielczością 0,9 eV. Emisję fotoelektronów wzbudzano za pomocą achromatycznej, dwuanodowej lampy rentgenowskiej, wyposażonej w anody: glinową oraz magnezową. Energia promieniowania wzbudzającego dla anody glinowej wynosi
1486,6 eV, natomiast dla anody magnezowej – 1253,6 eV.
Otrzymywane widma rozkładu elektronów wyemitowanych z powierzchni rejestrowane były w postaci cyfrowej, a następnie analizowane przy pomocy oprogramowania CasaXPS. Typowa procedura obróbki widma szczegółowego obejmowała: odejmowanie tła oraz procedurę dopasowania linii modelowych do wyników eksperymentalnych. Identyfikacja stanów chemicznych odpowiadających położeniom odpowiednich linii spektralnych opierała się na informacjach zawartych w literaturze przedmiotu oraz w katalogu widm XPS [123].
5.6 Spektrofotometria w zakresie nadfioletu i promieniowania widzialnego
(UV-Vis)
Analizę ilościową badanych roztworów wodnych wybranych zanieczyszczeń organicznych wykonano metodą spektrofotometrii UV-Vis, opartej na pomiarze absorbancji Aλ badanego roztworu, przy analitycznej długości fali. Analizę
przeprowadzono za pomocą spektrofotometru Jasco V-630, Japonia.
36
5.6.1 Pomiar adsorpcji zanieczyszczeń organicznych z roztworów wodnych na otrzymanych węglach aktywnych
W kolbach Erlenmayera umieszczono naważki adsorbentu (0 – 50 mg), do których dodano 100 cm3 wodnego roztworu fenolu (100 mg/dm3) lub BR 18 (70 mg/dm3), lub RR 198 (30 mg/dm3). Proces adsorpcji prowadzony był przez 4 h, w temperaturze
30 °C w łaźni wodnej z wytrząsaniem. Stężenia poszczególnych filtratów po procesie adsorpcji na węglach aktywnych wyznaczone zostały spektrofotometrycznie w oparciu o równania krzywych wzorcowych wykreślonych dla poszczególnych adsorbatów (Tabela 6).![]()
Tabela 6. Równania krzywych kalibracji wyznaczonych dla poszczególnych adsorbatów
Adsorbat | Równanie krzywej kalibracji | Współczynnik dopasowania R2 | Stężenie zanieczyszczenia w roztworze wodnym |
Fenol | Abs = 0,0151 ∙ Ct | 1,0000 | Ct = Abs/0,0151 |
BR 18 | Abs = 0,0463 ∙ Ct | 0,9995 | Ct = Abs/0,0463 |
RR 198 | Abs = 0,0142 ∙ Ct | 0,9999 | Ct = Abs/0,0142 |
37
6. Wyniki i dyskusja
6.1. Wyznaczenie warunków karbonizacji wyjściowej mieszaniny
W celu określenia warunków obróbki termicznej surowych mieszanin (związek magnezu/PET), wykonano szereg analiz termograwimetrycznych, zarówno poszczególnych związków magnezu jak i ich mieszanin z PET (Tabela 7). Analizy TG przeprowadzono w atmosferze argonu, w 850 °C. Czas ogrzewania w temperaturze docelowej wynosił 1 godzinę.
Tabela 7. Wykaz materiałów poddanych analizie termograwimetrycznej
![]()
Materiał Skład mieszaniny
[%wag. zw. Mg/ %wag. PET]
![]()
PET 0/100
![]()
3MgCO3·Mg(OH)2·3H2O/PET
100/0
30/70
50/50
70/30
Mg(OH)2/PET
![]()
100/0
30/70
50/50
70/30
MgO/PET
![]()
30/70
50/50
70/30
![]()
6.1.1 Analiza TG PET
Poli(tereftalan etylenu) użyty, jako prekursor węgla w materiałach wyjściowych
ulegał jednoetapowemu rozkładowi (Rysunek 6), w zakresie temperatur od 390 °C do
490 °C. Ubytek masy podczas zwęglania czystego PET wyniósł około 81% [124].
38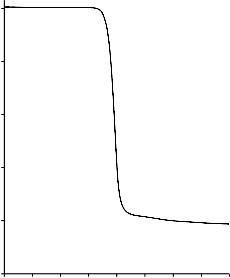
100
80
60
40
20 PET
0
50 150 250 350 450 550 650 750 850
Temperatura/oC
Rysunek 6. Termogram dla czystego poli(teraftalanu etylenu)
6.1.2 Analiza TG i XRD związków nieorganicznych oraz ich mieszanin z PET po karbonizacji
Wodorotlenek magnezu
Analiza termograwimetryczna czystego wodorotlenku magnezu potwierdziła jednoetapowy rozkład (ubytek masy próbki ok. 30%), w zakresie temperatur 300 –
390 °C [125]. Podczas ogrzewania Mg(OH)2 następuje jego rozkład do MgO
z wydzieleniem H2O:
Mg(OH)2→ MgO + H2 O (300 – 390 °C) (4)
Termogramy mieszanin Mg(OH)2/PET charakteryzowały się dwuetapowy ubytkiem masy o różnej wielkości i intensywności:
- pierwszy niewielki w zakresie temperaturze od 360 °C do 400 °C (ubytek masy 3 –
5%);
- drugi bardziej wyraźny w temperaturze od 560 °C do 650 °C (ubytek masy 25 – 40%).
39
100
90
80
czysty Mg(OH)2
70
60
70/30
50/50
30/70
50
40
30
50 150 250 350 450 550 650 750 850
Temperatura [°C]
Rysunek 7. Termogram dla czystego Mg(OH)2 oraz mieszaniny wyjściowej Mg(OH)2/PET (30/70, 50/50, 70/30)
Obecność stałego produktu rozkładu mieszaniny wodorotlenku magnezu z PET, potwierdzono analizą składu fazowego (XRD). Na podstawie uzyskanego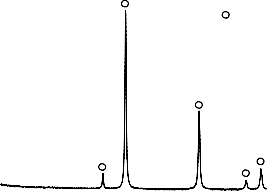
dyfraktogramu potwierdzono obecność fazy krystalicznej w postaci tlenku magnezu.
MgO
![]()
10 20 30 40 50 60 70 80
2θ [°]
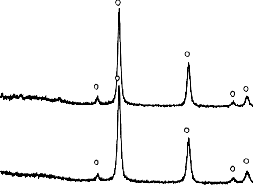
Rysunek 8. Dyfraktogram XRD przykładowego materiału węglowego otrzymanego
z mieszaniny Mg(OH)2/PET kalcynowanego w temperaturze 550 °C
40
Lekki zasadowy węglan magnezu (BMC)
Na podstawie przebiegu krzywej TG czystego BMC (Rysunek 9) można wyróżnić trzy przedziały spadku masy w zakresie temperatur od 266 °C do 534 °C, z całkowitą stratą masy ok. 55%. Stałym końcowym produktem rozkładu BMC jest MgO [126]:
3MgCO3 + Mg(OH)2 ∙ 3H2O → 3MgCO3 ∙Mg(OH)2 + 3H2O (210 – 320 °C) (5)
3MgCO3 ∙Mg(OH)2 → 3MgCO3 + MgO + H2O (340 – 490 °C) (6)
3MgCO3 → 3MgO + 3CO2 (490 – 550 °C) (7)
W trakcie analizy termograwimetrycznej mieszaniny BMC/PET można było zaobserwować wyraźny spadek masy zaczynający się w temperaturze około 380 °C, a kończący się w temperaturze 620 °C (Rysunek 9). Brak ubytku masy w temperaturze około 250 °C, widocznego przy rozkładzie czystego BMC, przypisano częściowemu rozkładowi wyjściowej mieszaniny na etapie wstępnego ogrzewania surowej mieszaniny w 265 °C (patrz preparatyka).
41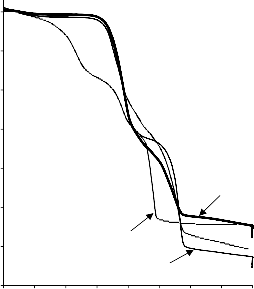
100
90
80
70
60
70/30
50
czysty BMC
40
50/50
30/70
30
50 150 250 350 450 550 650 750 850
Temperatura [°C]
Rysunek 9. Termogram dla czystego BMC oraz mieszaniny wyjściowej BMC/PET (30/70,
50/50, 70/30)
Analiza składu fazowego materiału węglowego (BMC/PET) po procesie karbonizacji w 550 °C
i 700 °C w skazała na całkowity rozkład związku nieorganicznego do MgO (Rysunek 10).
![]()
MgO
550 °C
700 °C
![]()
10 20 30 40 50 60 70 80
2θ [°]
Rysunek 10. Dyfraktogramy XRD materiałów węglowych otrzymanych z mieszaniny
BMC/PET 50/50 kalcynowanych w temperaturze 550 °C i 700 °C
42
Należy również zaznaczyć, że w przypadku wszystkich prezentowanych krzywych TG uzyskanych dla mieszanin PET z poszczególnymi związkami magnezu (Rysunek 7, Rysunek 9) odnotowano nieznaczne ubytki w masach próbek analizowanych mieszanin, powstałe w wyniku ich dalszego wygrzewania, w temperaturze powyżej 650 °C. Dalszy opis wyjaśniający przyczyny tego zjawiska został zawarty w rozdziale 7.4 niniejszej pracy, traktującym o mechanizmie powstawania porów w badanych sorbentach węglowych.
Tlenek magnezu
Termogram mieszaniny MgO/PET (Rysunek 11) wykazał jednoetapowy ubytek masy w zakresie temperatur 550 °C – 650 °C, związany z rozkładem PET. Powyżej
temperatury 650 °C nie stwierdzono znacznych zmian masy próbek.
100
90
70/30
80
50/50
70 30/70
60
50
40
30
50 150 250 350 450 550 650 750 850
Temperatura [°C]
Rysunek 11. Termogram mieszaniny wyjściowej MgO/PET (30/70, 50/50, 70/30)
Analiza składu fazowego mieszaniny MgO/PET 50/50 kalcynowanej w temperaturze
850 °C potwierdziła obecność w materiale węglowym tlenku magnezu (Rysunek 12).
43![]()
MgO

![]()
10 30 50 70
2θ [°]
Rysunek 12. Dyfraktogramy XRD materiału węglowego otrzymanego z mieszaniny
MgO/PET kalcynowanej w temperaturze 850 °C
Temperatury karbonizacji surowych mieszanin dobrano na podstawie analizy termogramów (Rysunek 6, Rysunek 7, Rysunek 9, Rysunek 11). Mając na względzie, przebieg krzywych TG polimeru (Rysunek 6) i czystych związków magnezu (Rysunek 7, Rysunek 9), jako minimalną temperaturę potrzebną do całkowitego ich rozkładu, wytypowano 550 °C. Jednakże, w przypadku wygrzewania mieszanin wyjściowych przeprowadzenie rozkładu ich komponentów wymagało zastosowania wyższych temperatur: 700 °C i 850 °C.
6.2 Charakterystyka otrzymanych materiałów węglowych
6.2.1 Pomiar izoterm adsorpcji/desorpcji N2 w 77 K wybranych materiałów węglowych
Zgodnie z klasyfikacją IUPAC [127] otrzymane doświadczalnie izotermy adsorpcji/desorpcji azotu w 77 K dla badanych materiałów węglowych oraz komercyjnego węgla aktywnego (Rysunki 13 – 16) w początkowym zakresie ciśnienia względnego p/p0 odpowiadały typowi I, a w zakresie średnich i wyższych typowi IV. Wzrost adsorpcji w zakresie niskich wartości ciśnienia względnego wskazuje na mikroporowaty charakter materiałów. Natomiast cechą charakterystyczną izoterm typu IV jest wystąpienie wyraźnie wykształconej pętli histerezy, związanej ze zjawiskiem kondensacji kapilarnej w obszarze mezoporów i limitowanej zdolności adsorpcyjnej
w wysokim obszarze ciśnień względnych p/p0 [14].
44
b) 600
12
PET 850°C
10
500
CWZ-35
400
8
300
6
4 200
2 100
0
0.0 0.3 0.5 0.8 1.0
p/p0
0
0 0,2 0,4 0,6 0,8 1
p0/p
Rysunek 13. Izotermy adsorpcji/desorpcji N2 w 77 K: a) karbonizatu z PET; b)
komercyjnego węgla CWZ-35
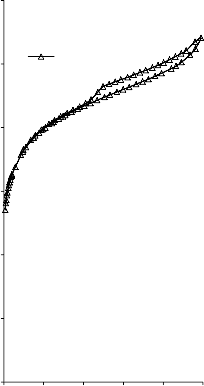
Kształt izotermy adsorpcji karbonizatu otrzymanego po pirolizie PET wskazuje na mikroporowaty charakter materiału. Jednakże, biorąc pod uwagę znikomą ilość zaadsorbowanego azotu, materiał ten można określić, jako nieporowaty.
45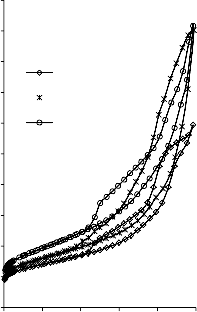
a) 2000
b) 2000
1800
1600
Mg(OH)2/PET
30/70
1800
1600
Mg(OH)2/PET
70/30
1400 50/50
1400
550 °C
700 °C
1200
1000
70/30
1200
1000
850 °C
800
800
600
600
400
400
200
200
0
0 0,2 0,4 0,6 0,8 1
p/po
0
0 0,2 0,4 0,6 0,8 1
p/po
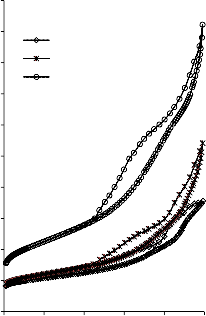
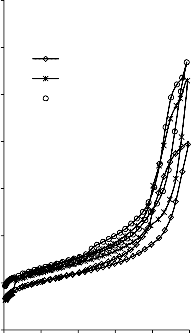
Rysunek 14. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych z mieszaniny: a) Mg(OH)2/PET (30,70, 50/50, 70/30) w temperaturze 850 °C; b) Mg(OH)2/PET (70/30) w zakresie temperatur 550 °C - 850 °C
25a0)0

16b0) 0
2250
2000
1750
30/70 BMC/PET
50/50 850 °C
70/30
550 °C
1400 700 °C
850 °C
1200
BMC/PET
50/50
1500
1000
1250
800
1000
600
750
500
250
400
200
0
0 0,2 0,4 p/p
0,6 0,8 1
0
0 0,2 0,4 0,6 0,8 1 p/p0
Rysunek 15. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych z mieszaniny: a) BMC/PET (30,70, 50/50, 70/30) w temperaturze 850 °C; b) BMC/PET (50/50) w zakresie temperatur 550 °C – 850 °C
46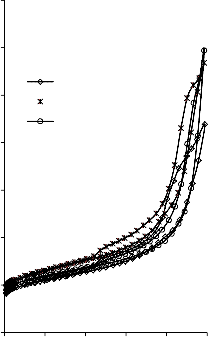
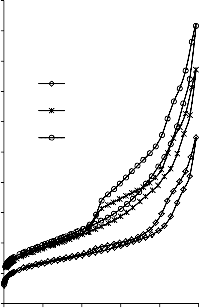
a) 1400
b) 1400
1200
30/70
MgO/PET
1200
550 °C
700 °C
MgO/PET
50/50
1000 50/50
70/30
800
850 °C
1000 850 °C
800
600
600
400
400
200
200
0
0 0,2 0,4 0,6 0,8 1 p/p0
0
0 0,2 0,4 0,6 0,8 1 p/p0
Rysunek 16. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych z mieszaniny: a) MgO/PET (30,70, 50/50, 70/30) w temperaturze 850 °C; b) MgO/PET (50/50) w zakresie temperatur 550 °C – 850 °C
W oparciu o doświadczalne niskotemperaturowe izotermy adsorpcji/desorpcji N2 badanych węgli aktywnych wyznaczono parametry charakteryzujące ich strukturę porowatą (Tabela 8).
47
Tabela 8. Parametry strukturalne materiałów węglowych wyznaczone na podstawie niskotemperaturowych izoterm adsorpcji/desorpcji azotu
![]()
Udział składnika
[%wag.]
Temperatura karbonizacji
![]()
[°C]
SBET
[m2/g]
Stotal
[m2/g]
![]()
Metoda αs
Sext
[m2/g]
Smicro
[m2/g]
Vtot 0,95
[cm3/g]
Vmicro DR
[cm3/g]
Vmeso
[cm3/g]
![]()
PET
Mg(OH)2/PET
0/100 850 34 37 3 34 0,02 0,02 0
30/70 850 907 1008 622 385 1,70 0,38 1,32
50/50 850 1049 1124 776 348 2,23 0,43 1,80
550 875 836 643 193 1,34 0,36 0,98
70/30
700 1253 1259 1029 239 1,92 0,51 1,41
![]()
850 1302 1317 1019 298 2,03 0,52 1,51
30/70 850 944 1088 470 618 1,25 0,39 0,86
550 531 519 284 235 0,73 0,21 0,52
BMC/PET
50/50
700 1064 1150 677 473 1,39 0,42 0,97
850 1049 1135 632 503 1,64 0,43 1,21
70/30 850 1984 2063 1505 557 2,93 0,79 2,14
![]()
30/70 850 713 882 390 492 1,12 0,32 0,80
550 678 722 381 341 1,04 0,30 0,74
MgO/PET
50/50
700 799 915 332 584 1,27 0,35 0,92
850 788 916 343 573 1,57 0,37 1,20
![]()
70/30 850 763 958 379 579 1,28 0,35 0,93
![]()
CWZ-35 - - 1260 1494 335 1159 0,81 0,61 0,2
48
6.2.2 Wpływ temperatury karbonizacji wyjściowych mieszanin oraz ich składy, na
rozwój porowatości w otrzymanych materiałach węglowych
Analizując wykresy izoterm adsorpcji/desorpcji azotu poszczególnych materiałów węglowych, można zauważyć tendencję wzrostową adsorpcji azotu wraz z zwiększeniem zawartości związków magnezu stosowanych do otrzymania mieszanin wyjściowych z PET (Rysunek 14 a, Rysunek 15 a, Rysunek 16 a). Istotną rolę w zmianach stopnia rozwinięcia struktury porowatej badanych węgli aktywnych odegrała również temperatura karbonizacji wyjściowej mieszaniny (Tabela 8). Tendencja ta jest szczególnie wyraźna dla węgli karbonizowanych w 550n°C i 850°C. Na podstawie przedstawionych wyników można stwierdzić, że w obszarze niskich ciśnień względnych izotermy adsorpcji azotu materiałów węglowych otrzymanych w temperaturze 700 °C i 850 °C niemal pokrywały się.
Węgiel pirolityczny otrzymany z PET, bez dodatku związku magnezu w mieszanie wyjściowej charakteryzuje się słabo rozwiniętą porowatością, z niewielkim udziałem mikroporów. Dodatek związku magnezu w mieszaninie z PET wpłynął znacząco na rozwinięcie struktury porowatej materiałów węglowych. Niezależnie od użytego związku magnezu i temperatury procesu, wszystkie badane adsorbenty były materiałami mikro- i mezoporowatymi, o silnie rozwiniętych powierzchniach.
Wyznaczona powierzchnia właściwa zmieniała się w przedziale od ok. 500 m2/g
(BMC/PET 50/50 550 °C) do około 2000 m2/g (BMC/PET 70/30 850 °C). Również całkowita objętość porów (Vtotal 0,95) węgli aktywnych osiągnęła duże wartości w przedziale od ok. 0,70 cm3/g do około 3,00 cm3/g.
Analizując parametry struktury porowatej materiałów węglowych otrzymanych z mieszaniny BMC/PET oraz Mg(OH)2/PET należy stwierdzić, że skutkiem zwiększenia ilości związku magnezu w mieszaninie wyjściowej był wyraźny wzrost powierzchni całkowitej oraz udziału mezoporów w strukturze badanych adsorbentów. Ponadto, zaobserwowano istotny rozwój struktury porowatej badanych węgli wraz z przyrostem temperatury karbonizacji. Szczególnie wyraźny wzrost powierzchni i objętości mezoporów jest widoczny w przypadku węgli aktywnych otrzymanych z mieszaniny Mg(OH)2/PET 70/30, karbonizowanej w zakresie temperatur od 550 °C do 850 °C (Tabela 8). Warto również podkreślić, że temperatura karbonizacji wpłynęła znacząco na poprawę struktury porowatej w obszarze mikroporów. Świadczą o tym
wartości Smicro i Vmicro DR, które zmieniały się w szerokim przedziale (Mg(OH)2/PET
49
70/30 550 °C – 850 °C: Smicro 193 m2/g – 298 m2/g, Vmicro DR 0,36 cm3/g – 0,52 cm3/g; BMC/PET 50/50 550 °C – 850 °C: Smicro 235 m2/g – 503 m2/g, Vmicro DR 0,21 cm3/g –
0,43 cm3/g).
Materiały węglowe otrzymane z mieszaniny MgO/PET charakteryzują się większym udziałem mikroporów, w porównaniu z węglami zawierającymi w mieszaninie wyjściowej Mg(OH)2 lub BMC. Zwiększenie udziału MgO w mieszanie wyjściowej oraz wzrost temperatury karbonizacji nie wpłynął tak znacząco na rozwój parametrów struktury porowatej zarówno w obszarze mikro-, jak i mezoporów otrzymanych materiałów (Tabela 8), jak to można było zaobserwować w przypadku dwóch pozostałych układów.
Należy mieć na względzie, że oprócz prekursora tlenku magnezu, na rozwój porowatości materiałów węglowych wpływa także rodzaj użytego prekursora węgla [61]. Ponadto, także znaczącą rolę w rozwoju struktury porowatej materiału odgrywa szybkość ogrzewania surowej mieszaniny. Dowiedziono, że przeprowadzenie karbonizacji z szybkością grzania 10 °C/min, pozwala na uzyskanie materiałów węglowych z większą zawartością mikroporów, w przeciwieństwie do szybkość grzania
5 °C/min [61].
Biorąc pod uwagę powyższą analizę wyników, można stwierdzić, że porowatość otrzymanych węgli aktywnych silnie zależy od składu jakościowego i ilościowego mieszaniny wyjściowej oraz temperatury karbonizacji. Wyraźny rozwój porowatości materiałów węglowych następował wraz ze zwiększeniem udziału wagowego związku magnezu w mieszanie wyjściowej. Adsorbenty otrzymane z mieszanin zawierających w składzie wyjściowym związek magnezu nieulegający rozkładowi termicznemu charakteryzowały się silnie rozwiniętą strukturą porowatą. Natomiast materiały otrzymane z mieszaniny wyjściowej zawierającej w swoim składzie MgO charakteryzowały się słabiej rozwiniętą strukturę porowatą. Temperatura karbonizacji istotnie wpłynęła na rozwoju struktury porowatej węgli. Materiały węglowe preparowane w najniższej temperaturze (550 °C) charakteryzowały się najsłabiej rozwiniętymi parametrami strukturalnymi; wraz ze wzrostem temperatury parametry te wyraźnie wzrastały.
50
6.2.3 Rozkład objętości porów w wybranych materiałach węglowych
W celu zbadania zależności pomiędzy rozmiarem porów badanych materiałów, a składem jakościowym i ilościowym mieszanin wyjściowych oraz temperaturą procesu otrzymywania, wykonano analizę rozkładu objętości porów wybranych węgli
aktywnych (Rysunek 17).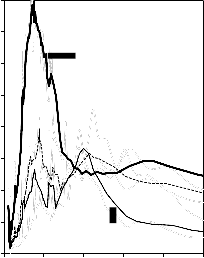
4,0
a)
3,5
3,0
BMC/PET, 850°C
70:30
4,0
b)
3,5
3,0
BMC/PET, 50:50
2,5
2,0
50:50
2,5
2,0
850°C
1,5
1,5
1,0
1,0
700°C
0,5
0,0
30:70
0 10 20 30 40 50
Szerokoś ć porów, nm
0,5
0,0
550°C
0 10 20 30 40 50
Szerokoś ć porów, nm

4,5
c)
4,0
3,5
3,0
Mg(OH)2/PET
50:50, 850 °C
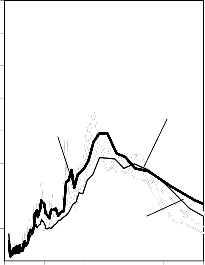
4,0
d)
3,5
3,0
2,5
MgO/PET
2,5
2,0
1,5
1,0
0,5
0,0
30:70, 850 °C
70:30, 850 °C
70:30, 550 °C
0 10 20 30 40 50
Szerokość porów [nm]
2,0
1,5
1,0
0,5
0,0
50:50, 850°C 50:50, 700°C
30:70, 700°C
0 10 20 30 40 50
Szerokość porów, nm
Rysunek 17. Funkcja rozkładu objętości porów wyznaczona metodą BJH dla węgli aktywnych otrzymanych z mieszaniny: a) BMC/PET (30/70, 50/50, 70/30) 850 °C, b) BMC/PET 50/50 (550 °C – 850 °C), c) Mg(OH)2/PET (30/70, 50/50, 70/30) 850 °C),
70/30 (550 °C), d) MgO/PET 30/70 (700 °C), 50/50 (700 °C, 850 °C)
51
Analiza rozkładu objętości porów badanych materiałów węglowych wskazuje na wyraźną zależność pomiędzy wymiarem mezoporów, a względnym udziałem związku magnezu w mieszaninie wyjściowej oraz temperaturą procesu. Maksimum adsorpcji w mezoporach uległo wyraźnemu wzrostowi wraz ze zwiększeniem ilości związku magnezu w mieszaninie wyjściowej z PET. Dotyczy to w szczególności mezoporów w zakresie od 5 nm do 12 nm, w układzie BMC/PET (Rysunek 17 a) oraz od 15 nm do
40 nm, w układzie Mg(OH)2/PET (Rysunek 17 c). Wzrost temperatury karbonizacji pociągnął za sobą zwiększenie wielkości porów. W przeciwieństwie do materiałów preparowanych w 550 °C, materiały otrzymane w temperaturze 700 °C i 850 °C wykazują wyraźny wzrost maksimum funkcji w zakresie mezoporów (Rysunek 17 b, Rysunek 17 c).
W przypadku materiałów węglowych otrzymanych z mieszaniny MgO/PET, udział MgO w mieszaninie wyjściowej oraz temperatura karbonizacji nie miały większego wpływu na wzrost maksimum funkcji rozkładu w obszarze mezoporów (Rysunek 17 d). Otrzymane adsorbenty charakteryzowały się znaczącym udziałem porów o wymiarach z zakresu od 20 nm do 40 nm, z wyraźnie mniejszym udziałem z przedziału od 5 do
12 nm.
Mając na względzie średnią wielkość krystalitów MgO (Tabela 9) wyznaczoną na podstawie uzyskanych dyfraktogramów (Rysunki 8, 10, 12) z równania Scherrera (Równanie 1), można zauważyć, że ich rozmiar w układzie BMC/PET (9 – 11 nm) i Mg(OH)2/PET (15 – 16 nm) pokrywa się z rozmiarem mezoporów i rośnie wraz z temperaturą karbonizacji mieszaniny wyjściowej.
Tabela 9. Średnia wielkość krystalitów MgO materiałów węglowych otrzymanych
z mieszaniny BMC/PET i MgO/PET karbonizowanych w różnym zakresie temperatur
![]()
BMC/PET (50/50) MgO/PET (50/50) Mg(OH)2/PET (50/50)
![]()
Średni
Temperatura
[°C]
Średni rozmiar krystalitu [nm]
Temperatura
[°C]
Średni rozmiar krystalitu [nm]
Temperatura
[°C]
rozmiar krystalitu [nm]
![]()
550 9
700 9
![]()
850 11
52
Natomiast, w przypadku układu MgO/PET niezależnie od temperatury wygrzewania,
średnia wielkość krystalitów MgO nie ulegała zmianie.
Zależność pomiędzy rozmiarem mezoporów, a wielkością krystalitów MgO potwierdził również Inagaki i in. [62]. Dowiedziono, że rozmiar mezoporów w materiale węglowym jest uzależniony od prekursora tlenku magnezu i pokrywa się z jego wielkością krystalitów.
6.3 Mechanizm tworzenia porów w otrzymanych materiałach węglowych
W trakcie procesu karbonizacji surowych mieszanin następuje rozkład PET oraz użytego związku magnezu. Na podstawie przeglądu literatury można oczekiwać, że zastosowanie związków ulegających rozkładowi daje dwojaki efekt, tj. generowanie mikroporów (działanie wydzielających się, CO2 i/lub H2O na materiał węglowy) oraz tworzenie mezoporów (działanie stałego produktu rozkładu na materiał węglowy) [61, 62]. Z kolei oczekuje się, że użycie związku nieorganicznego nieulegającemu rozkładowi (MgO) wpływa na preferencyjne tworzenie się mezoporów w otrzymywanym adsorbencie węglowym.
Z analizy dotychczas przedstawionych wyników, można zauważyć duże zróżnicowanie w strukturze porowatej, pomiędzy materiałami otrzymanymi z mieszaniny wyjściowej z PET zawierającej związek niestabilny termicznie tj. BMC lub Mg(OH)2, a nieulegającemu rozkładowi MgO (Tabela 8). Ponadto warunki otrzymywania materiału węglowego, odegrały także znaczącą rolę. Materiały węglowe otrzymane w 550 °C, charakteryzowały się najsłabiej rozwiniętymi parametrami strukturalnymi, wyraźnie wzrastającymi z temperaturą pirolizy.
Należy także zwrócić uwagę na wyniki pomiarów termograwimetrycznych czystych materiałów oraz ich mieszanin z PET. Niezależnie od składu jakościowego i ilościowego, rozkład mieszanin (związek magnezu/PET) kończył się w temperaturze powyżej 620 °C, przekraczającej punkt rozkładu poszczególnych komponentów mieszanin wyjściowych. W przypadku materiałów otrzymanych z mieszaniny BMC/PET, Mg(OH)2/PET, po przekroczenia temperatury 620 °C masa próbki uległa
wyraźnemu zmniejszeniu (Rysunek 7, Rysunek 18).
53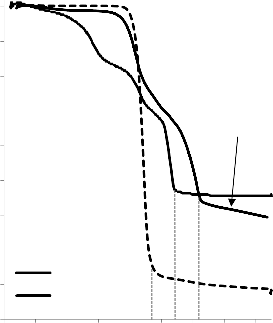
100
90
80
70 Ubytek w masie
węgla
60
50
40
30 PET
BMC
20
PET + BMC (50:50)
10
0 100 200 300 400 500 600 700 800
Temperatura [oC]
Rysunek 18. Krzywe TG PET, BMC oraz mieszaniny BMC/PET 50/50
Powyższe obserwacje, mogą świadczyć o zachodzącej reakcji pomiędzy gazami wydzielającymi się z niestabilnie termicznych związków magnezu, a materiałem węglowym tworzącym się z PET, powyżej temperatury 620 °C (Reakcja 1, Reakcja 2). W celu potwierdzenia tej możliwości, przeprowadzono analizę TPD dla poszczególnych mieszanin, uprzednio wygrzanych w temperaturze 700 °C (Rysunek 19, Rysunek 20, Rysunek 21). W trakcie pomiaru rejestrowano sygnały o stosunkach masy do ładunku (m/z) równych: m/z = 44 (ditlenek węgla), m/z = 18 (woda), m/z = 28 (tlenek węgla) oraz m/z = 2 (wodór).
Podczas analizy TPD próbki BMC/PET 50/50 w zakresie temperatur od 60 °C do około
300 °C zaobserwowano intensywne sygnały potwierdzające wydzielanie się wody oraz dwutlenku węgla (Rysunek 19).
54
m/z=18
BMC/PET 50/50
m/z=44
m/z=2
m/z=28

0 200 400 600 800 1000
Temperatura [°C]
Rysunek 19. Analiza TPD dla materiału węglowego otrzymanego z mieszaniny BMC/PET
50/50 700 °C
Wraz ze wzrostem temperatury intensywność sygnałów (m/z = 18, m/z = 44) zaczynała stopniowo maleć. Natomiast przyrostowi temperatury towarzyszył sukcesywny wzrost sygnałów pochodzących od CO i H2, z wyraźnym efektem powyżej 600 °C.
W przypadku materiałów węglowych otrzymanych z mieszaniny Mg(OH)2/PET,
zarejestrowano sygnał pochodzący od H2O, z największą intensywnością w zakresie temperatur od 100 °C do 300 °C. Intensywność wydzielającego się CO2 była wyraźnie niższa, w porównaniu z sygnałem pochodzącym od wody. Podobnie jak w przypadku mieszaniny BMC/PET, wraz ze wzrostem temperatury intensywność sygnałów ulegała osłabieniu, z wyraźnym spadkiem dla H2O w ~ 800 °C i CO2 w ~ 400 °C. Początkowo bardzo słabe sygnały pochodzące od wydzielającego się H2 i CO zaczynały zwiększać swoją intensywność wraz z przyrostem temperatury, z wyraźny efektem w 400 °C (m/z = 2) i 650 °C (m/z = 28). Ponadto, ilość wydzielających się gazów wzrastała wraz ze zwiększaniem zawartości wodorotlenku magnezu w mieszanie wyjściowej (Mg(OH)2/PET 70/30) (Rysunek 20).
55
Mg(OH)2/PET 30/70
Mg(OH)2/PET 70/30
m/z=2
m/z=18
m/z=2
m/z=44
m/z=44
m/z=18
m/z=28
m/z=28
0 200 400 600 | 800 | 1000 | 0 | 200 | 400 600 | 800 | 1000 |
Temperatura [°C] | Temperatura [°C] |


Rysunek 20. Analiza TPD dla materiałów węglowych otrzymanych z mieszanin:
a) Mg(OH)2/PET 30/70, b) Mg(OH)2/PET 70/30 700 °C
W przeciwieństwie do pozostałych materiałów, analiza TPD materiału węglowego - MgO/PET 50/50 (Rysunek 21) wykazała jedynie śladowe ilości wydzielającego się CO2, H2O oraz CO. Ponadto, w temperaturze ~ 600 °C został zarejestrowany sygnał pochodzący od H2.
m/z=18
m/z=2

0 200 400 600 800 1000
Temperatura [°C]
Rysunek 21. Analiza TPD dla materiału węglowego otrzymanego z mieszaniny MgO/PET
50/50 po pirolizie w 700 °C
56
Biorąc pod uwagę:
- intensywne wydzielanie się tlenku węgla i wodoru w temperaturze powyżej 600 °C, w materiałach węglowych zawierających w mieszaninie wyjściowej BMC lub Mg(OH)2;
- śladowe ilości wydzielającego się tlenku węgla i wodoru, w przypadku analizy
materiału węglowego otrzymanego z mieszaniny MgO/PET;
założono zajście procesu aktywacji, materiału węglowego utworzonego z PET, a gazowymi produktami rozkładu BMC lub Mg(OH)2, pełniącymi rolę czynników aktywujących.
Wyraźnie mniejszy ubytek masy węgla podczas rozkładu mieszanin, w porównaniu z rozkładem czystego PET, może być wynikiem okluzji w porach części gazowych produktów rozkładu poszczególnych komponentów. Część gazów wydzielających się z niestabilnie termicznie związków magnezu, jest zaokludowana w formowanych porach do czasu, gdy temperatura osiągnie ~ 700 °C, w tej temperaturze następuję gazyfikacja węgla formowanego z PET przez CO2 lub/i H2O według reakcji (1) i (2).
W wyniku gazyfikacji węgla następuje rozwinięcie struktury porowatej materiału,
z wytworzeniem jak największej ilości przypadkowo rozłożonych porów różnych rozmiarów (Rysunek 22).
57
CO2 i/lub H2O
- opuszczające powierzchnie
skarbonizowanego PET
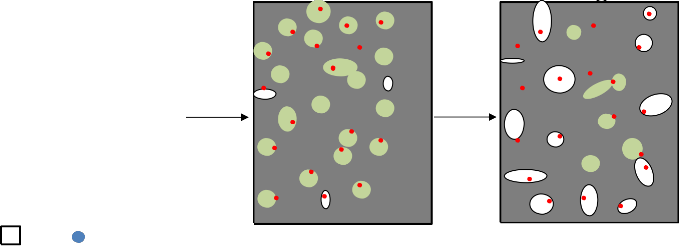
![]()
- okluzja gazowych produktów rozkładu związku magnezu w porach węgla formowanego z PET
gazyfikacja węgla przez CO2 i/lub H2O
- rozwinięcie struktury porowatej
w wyniku otwarcia porów
ogrzewanie ogrzewanie
do ~ 600 °C
od ~ 700 °C
PET + BMC lub Mg(OH)2
MgO
CO2 i/lub H2O
Rysunek 22. Proponowany mechanizm tworzenia porów w materiale węglowym
58
Intensywne wydzielanie się CO2, H2O, CO i H2 (Rysunek 19, Rysunek 20), może być także powiązane z rozkładem tlenowych grup funkcyjnych, obecnych na powierzchni węgla. Zawierające tlen grupy powierzchniowe rozkładają się w różnych zakresach temperatur, tworząc lotne produkty gazowe. Ditlenek węgla może się wydzielać w wyniku rozkładu grup karboksylowych i laktonowych w temperaturze
350 °C – 700 °C, tlenek węgla na skutek rozkładu grup chinonowych i fenolowych w zakresie 500 °C – 950 °C, a para wodna pochodzi z grup karboksylowych i fenolowych w zakresie temperatur 200 °C – 600 °C [14]. W celu powiązania jakości wydzielających się gazów z rozkładem grup funkcyjnych obecnych na powierzchni węgla wykonano analizę XPS (Tabela 10), na dwóch przykładowych materiałach węglowych otrzymanych z mieszaniny BMC/PET oraz MgO/PET.
Tabela 10. Dekonwolucja pasma C (1s) w widmie XPS dla materiałów węglowych
otrzymanych z mieszaniny BMC/PET, MgO/PET
Położenie pasma C (1s) [eV] | |||||
Próbka | 284,6 frakcje grafitowego węgla | 286,0 węgiel z C-O (fenolowe, eterowe grupy) | 287,2 węgiel C=O (karbonylowe, chinynowe grupy) | 288,7 węgiel z O-C=O (karboksylowe lub estrowe grupy) | 290,2 węgiel związany z CO2 |
BMC/PET | 72,0 | 12,9 | 6,0 | 3,4 | 5,7 |
MgO/PET | 78,0 | 10,5 | 4,8 | 2,9 | 3,8 |
Pojawiający się sygnał w paśmie węgla C (1s) dla obu materiałów wykazuje asymetryczne ogonowanie (Rysunek 23), które związane są z obecnością tlenowych
grup funkcyjnych.
59
a)
294
292
290
288
286
284
282
280
Energia wiązania [eV]
b)
294
292
290
288
286
284
282
280
Energia wiązania [eV]
Rysunek 23. Dekonwolucja pasma C1s w widmie XPS dla materiałów węglowych
otrzymanych z mieszaniny: a) BMC/PET, b) MgO/PET
Pasmo C (1s) widma XPS potwierdziło obecność grup fenolowych i eterowych, którym
odpowiada pik przy 286,0 eV, karboksylowych lub estrowych dających pik przy
288,7 eV i grupy karbonylowych lub chinonowych dających pik przy 287,2 eV (Tabela
10). Frakcje grafitowego węgla i węgla związanego z CO2 dają pik przy 284,6 eV
i 290,2 eV.
Wartym podkreślenia jest to, że oba materiały węglowe wykazały zbliżoną zawartość tlenowych grup funkcyjnych. Na ich powierzchni ponadto, wszystkie oznaczane grupy powierzchniowe (Tabela 10) są termicznie stabilnie w temperaturze poniżej 200 °C [14,
128, 129]. Jak pokazano w Tabeli 10, zawartość powierzchniowych kompleksów (grupy
karbonylowa, fenolowa i eterowa) ulegających rozkładowi z wydzieleniem CO, w wyższych temperaturach [129] jest porównywalna w obu materiałach. Co więcej,
60
analiza TPD potwierdza pojawienie się intensywnych sygnałów m/z = 2 oraz m/z = 28, jedynie w przypadku materiałów otrzymanych z mieszanin wyjściowych, zawierających niestabilne termicznie związki magnezu (BMC, Mg(OH)2). Ponadto, podczas generowania gazów towarzyszy ubytek węgla (Rysunek 7, Rysunek18). Potwierdza to zachodzenie reakcji gazyfikacji węgla wydzielającymi się CO2 i H2O, w wyniku której następuje wydzielenie się H2 i CO, w wyższych zakresach temperatur ( ~700 °C). Oczywistym jest również, że niewielka ilość CO i H2 wydziela się w trakcie otrzymywania materiału do analizy TPD (700 °C). W konsekwencji, dochodzi do częściowej okluzji powyższych gazów wraz z CO2 i/lub H2O (BMC, Mg(OH)2), w formowanym węglu, a następnie do ich uwolnienia w temperaturze powyżej 600 °C (Rysunek 19, Rysunek 20). Jak wspomniano, w trakcie analizy TPD materiału węglowego MgO/PET zarejestrowano niewielkie sygnały pochodzące od wody oraz wodoru (Rysunek 21). Nieznaczny wzrost sygnału m/z = 18 zauważalny do temperatury około 200 °C może wynikać z zawilgocenia próbki. Natomiast sygnał pochodzący od wodoru (m/z=2) widoczny w temperaturze powyżej 600 °C może wynikać z fragmentacji produktów rozkładu PET [130]. Tak, więc proces gazyfikacji węgla tworzącego się podczas ogrzewania MgO zmieszanego z PET, nie ma miejsca. W rezultacie, niezależnie od temperatury, skumulowana objętość porów obliczana dla węgli uzyskanych z mieszaniny MgO/PET jest stosunkowo niska.
6.4 Wpływ domieszek na wydajność tworzenia węgla w układach związek
magnezu/PET
Efektem aktywowania materiału węglowego jest kreowanie porów, a tym samym znaczny wzrost powierzchni właściwej. Jednakże procesowi temu nieodłącznie towarzyszy dodatkowy, często bardzo znaczny ubytek masy. Taka droga otrzymywania jest, zatem obarczona bardzo niską całkowitą wydajnością produktu, wynoszącą kilka procent, co w porównaniu z węglami aktywnymi otrzymywanymi z surowców konwencjonalnych jest bardzo niekorzystne i znacznie wpływa na obniżenie opłacalności produkcji. Ze względu na wyraźne zmniejszenie stopnia rozkładu mieszanin (związek magnezu/PET), przekraczającego punkt rozkładu dla poszczególnych komponentów mieszaniny wyjściowej (rozdział 6.4), przystąpiono do
zbadania wydajności węglowej poszczególnych układów. W każdym przypadku po
61
procesie pirolizy mieszaniny PET ze związkiem magnezu (BMC, Mg(OH)2, MgO) w materiale węglowym pozostaje stała pozostałość, w postaci tlenku magnezu. W celu określenia ilości pozostałości nieorganicznej (MgO) w otrzymanym materiale węglowym po procesie pirolizy, przeprowadzono dodatkowo analizę termograwimetryczną w atmosferze powietrza. Na podstawie różnicy mas otrzymanych po procesie analizy termograwimetrycznej w poszczególnych środowiskach, oszacowano wydajność węglową otrzymanych materiałów w temperaturze 700 °C
i 850°C (Tabela 11).
62
Tabela 11. Wydajność węglowa materiałów otrzymanych w wyniku ogrzewania mieszaniny PET z różnymi związkami magnezu
![]()
Mieszanina wyjściowa
Procent masowy, [%wag.]
Wydajność węglowa po wygrzewaniu do
700°C,
![]()
[%] (a)
Wydajność węglowa po zakończeniu procesu,
[%]
(b)
Zmiany
w wydajnośc i węglowej, [%]
(a – b)
Teoretycznie możliwa do osiągnięcia wydajność, [%]
MgO/PET
30/70 27,3 26,1 1,2 -
50/50 27,7 26,6 1,1 -
70/30 27,4 26,7 0,8 -
Mg(OH)2/PET
30/70 28,8 27,4 1,4 38,9
50/50 29,2 27,7 1,5 53,3
70/30 31,0 29,9 1,1 81,0
BMC/PET
30/70 38,1 35,1 3,0 45,3
50/50 40,6 37,8 2,8 64,2
![]()
70/30 49,0 45,1 3,9 88,4
Wydajność procesu otrzymywania poszczególnych materiałów otrzymanych z mieszaniny związek magnezu/PET oscyluje w granicach od około 27% w układzie MgO/PET, do prawie 50% w układzie BMC/PET 70/30 (Tabela 11). W przypadku materiałów otrzymanych z mieszanin Mg(OH)2/PET, BMC/PET można zauważyć, że wraz ze zwiększaniem udziału wagowego związku magnezu w mieszaninie wyjściowej, wydajność procesu rośnie. W przeciwieństwie do materiałów otrzymanych z mieszaniny wyjściowej zawierającej stabilnie termiczny związek magnezu (MgO), gdzie wartości wydajności węglowej są do siebie bardzo zbliżone. Różnice w wydajności węglowej pomiędzy uzyskanymi materiałami, mogą być związane z okluzją gazowych produktów rozkładu związku magnezu (CO2 i/lub H2O) w węglu formowanym z PET (patrz podrozdział 7.4)
Należy jednak wziąć pod uwagę, że uzyskane hipotetyczne wydajności węglowe materiałów, przy założeniu całkowitej okluzji gazów, mają o wiele wyższe wartości, w porównaniu z wartościami rzeczywistymi. Pozwala to przypuszczać, że zjawisko
okluzji ma jedynie charakter częściowy.
63
Proces gazyfikacji materiału węglowego zachodzi w temperaturze powyżej 600 °C, czego efektem jest intensywne wydzielanie się CO i H2, co musi być powiązane ze spadkiem masy węgla. Takie zachowanie możemy zauważyć w przypadku porównania dwóch typów materiałów wyjściowych. Węgle otrzymane z mieszaniny PET z BMC lub Mg(OH)2 wykazują o wiele wyższy spadek masy podczas procesu ogrzewania, powyżej temperatury 620 °C, w porównaniu z materiałem otrzymanym z mieszaniny wyjściowej zawierającej MgO. Dlatego też, zarówno okluzja gazowych produktów rozkładu użytych związków magnezu, jak i gazyfikacja materiału, mają znaczący wpływ na wydajność węglową otrzymanych materiałów węglowych. Niemniej jednak, poprzez zróżnicowany stopień gazyfikacji węgla, przez CO2 i H2O oraz częściową okluzję wymienionych gazów, ilościowe oszacowanie wpływu każdego czynnika jest obarczone dużym błędem. Oczywistym jest, że MgO nie będzie ulegał rozkładowi podczas procesu priolizy. Jednakże wydajność węglowa dla materiałów otrzymanych z mieszaniny MgO/PET jest wyższa (27%) niż dla karbonizatu PET (19%). W tym wypadku, przyczyną zwiększonej wydajności może być efekt utrudnionego płynięcia
termoplastycznego prekursora (PET) [62] i/lub zwiększone usieciowanie polienowej
struktury w obecności tlenków tj. tlenku magnezu [131]. Co więcej, nie powinno się wykluczać tego samego efektu w przypadku mieszaniny BMC/PET oraz Mg(OH)2/PET, gdyż proces pirolizy tych surowców prowadzi również do wytworzenia w końcowym etapie MgO.
Podsumowując, wartości wydajności węglowej zależą zarówno od jakościowego jak i ilościowego składu mieszaniny wyjściowej. Ponadto, wydajność przeprowadzonego procesu, w niektórych układach jest obarczona dużym błędem, w skutek częściowej okluzji gazowych produktów rozkładu użytych związków magnezu w mieszaninie wyjściowej z PET.
6.5 Zdolność adsorpcyjna materiałów węglowych względem wybranych
zanieczyszczeń organicznych
Ze względu na znaczną zawartość mikro- i mezoporów w otrzymanych materiałach węglowych, przeprowadzono na nich adsorpcję modelowych związków organicznych o różnych masach cząsteczkowych takich jak fenol, czerwień anilinowa
(BR 18) i czerwień reaktywna (RR 198).
64
6.5.1 Usuwanie fenolu z wody
W celu określenia przydatności otrzymanych materiałów węglowych do usuwania związków o małej masie cząsteczkowej, przeprowadzono badania adsorpcji roztworu fenolu (Rysunek 24, Rysunek 25). Wszystkie zbadane adsorbenty wykazują zdolność adsorpcyjną względem tego zanieczyszczenia, za wyjątkiem karbonizatu PET. Najmniejszą zdolność adsorpcyjną względem fenolu, spośród materiałów węglowych otrzymanych z mieszaniny związku magnezu z PET, wykazał Mg(OH)2/PET (Rysunek 24 b). Ponadto, niezależnie od udziału wagowego wodorotlenku magnezu w mieszaninie wyjściowej, stopień usunięcia zanieczyszczenia jest zbliżony. Wydajność adsorpcji zastosowanego zanieczyszczenia organicznego z wody była większa, w przypadku materiałów węglowych otrzymanych z mieszaniny BMC/PET oraz MgO/PET (Rysunek 24 c, Rysunek 24 d). Można tutaj także zauważyć, wyraźniejszy wpływ udziału wagowego związku magnezu w mieszaninie wyjściowej na zdolność adsorpcyjną roztworu fenolu. W porównaniu z materiałami otrzymanymi z mieszaniny związek magnezu/PET, komercyjny węgiel aktywny CWZ-35 (Rysunek
24 a) wykazał wyższą skuteczność usuwania użytego zanieczyszczenia.
65
a) 100
90
80
70
60
50
40
30
20
10
0

PET CWZ-35
b) 100
90
80
70
60
50
40
30
Mg(OH)2/PET
850 °C
30/70
50/50
70/30
c) 100
0 10 20 30 40 50
Dawka materiału węglowego [mg]
d)100

0 10 20 30 40 50
Dawka materiału węglowego[mg]
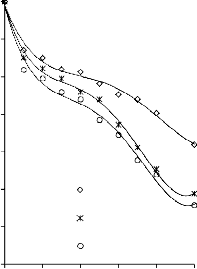
90 90
80 80
70 70
60 60
50 BMC/PET
850 °C
40
30
30/70
50/50
70/30
50 MgO/PET
850 °C
40
30
30/70
50/50
70/30
0 10 20 30 40 50
Dawka materiału węglowego [mg]
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 24. Adsorpcja fenolu z wodnego roztworu na a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny b) Mg(OH)2/PET, c) BMC/PET, d) MgO/PET
Adsorpcyjne usuwanie zanieczyszczeń wodnych za pomocą węgli aktywnych zależy od ich powierzchni właściwej oraz objętości i rozmiaru porów. W przypadku adsorpcji zastosowanego w badaniach roztworu fenolu, udział mikroporów w materiale węglowym odgrywa znaczącą rolę [104]. Jak wynika z danych przedstawionych w Tabeli 8, materiały węglowe otrzymane z mieszanin wyjściowych (Mg(OH)2/PET, BMC/PET, MgO/PET) karbonizowanych w 850 °C, charakteryzowały się zbliżonym udziałem mikroporów, z niewielką tendencją wzrostową powierzchni i objętości
mikroporów wraz z udziałem związku magnezu w mieszanie wyjściowej. Tendencja ta
66
przekłada się na zbliżoną zdolność adsorpcyjną materiałów węglowych względem roztworu fenolu (Rysunek 24). Niska zdolność adsorpcyjna karbonizatu PET względem fenolu, przekłada się na jego bardzo małą powierzchnią i objętością mikroporów (Tabela 8).
Skuteczność procesu adsorpcji roztworu fenolu na porowatych materiałach węglowych
rosła wraz z temperaturą karbonizacji mieszaniny wyjściowej (Rysunek 25).
a) 100
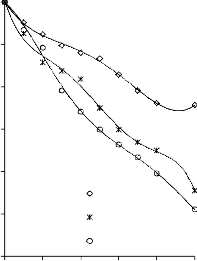
b) 100

90 90
80 80
70 70
60
Mg(OH)2/PET
50 70/30
40
550
700
850
60
BMC/PET
50 50/50
40
550
700
850
0 10 20 30 40 50
Dawka materiłu węglowego [mg]

c) 100
0 10 20 30 40 50
Dawka materiału węglowego [mg]
90
80
70
60
MgO/PET
50/50
50
40
550
700
850
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 25. Adsorpcja fenolu z wodnego roztworu na węglach aktywnych otrzymanych
w różnych temperaturach mieszaniny a) Mg(OH)2/PET, b) BMC/PET, c) MgO/PET
Materiały węglowe otrzymane w temperaturze 550 °C wykazały najmniejszy stopień usunięcia fenolu (Rysunek 25), w stosunku do materiałów otrzymanych w 700 i 850 °C.
67
Tendencja ta wynika, ze wzrostu powierzchni i objętości mikroporów wraz z temperaturą karbonizacji materiałów wyjściowych (Tabela 8).
6.5.2 Usuwanie barwników z wody
Skuteczność adsorpcji w dużym stopniu jest uzależniona od wielkości cząsteczek adsorbatu i struktury porowatej adsorbentu. Użyte w badaniach adsorbenty węglowe charakteryzowały się dużym objętością mezoporów (Tabela 8). W celu określenia zdolności adsorpcyjnej materiałów węglowych względem zanieczyszczeń o większej masie cząsteczkowej, przeprowadzono szereg pomiarów adsorpcji kationowego BR 18 (Rysunek 26, Rysunek 27) oraz anionowego RR 198 (Rysunek 28, Rysunek 29) barwnika.
Efektywność usuwania tych substancji organicznych zależała zarówno od udziału wagowego związku magnezu w mieszaninie wyjściowej (Rysunek 26, Rysunek 28) jak i od temperatury karbonizacji (Rysunek 27, Rysunek 29).
Materiały węglowe otrzymane z mieszaniny niestabilnego termicznie związku magnezu (Mg(OH)2, BMC) z PET charakteryzują się wiele wyższym stopniem usunięcia barwnika BR18 z roztworu (Rysunek 26 b, Rysunek 26 c) niż węgle aktywne otrzymane z mieszaniny MgO/PET (Rysunek 26 d). Ponadto, w przypadku materiałów otrzymanych z mieszaniny Mg(OH)2/PET, BMC/PET zauważalny jest wyraźny wpływ udziału związku magnezu w mieszaninie wyjściowej na proces adsorpcji roztworu
barwnika (Rysunek 26 b, Rysunek 26 c).
68
a) 70
60
50
b) 70
60
50
Mg(OH)2/PET
850 °C
30/70
50/50
70/30
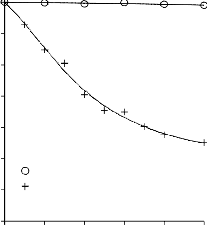
40 40
30 30
20 20
PET
10 CWZ-35 10
0
0 10 20 30 40 50

Dawka materiału węglowego [mg]
0
0 10 20 30 40 50

Dawka materiału węglowego [mg]
c) 70
60
50
BMC/PET
850 °C
30/70
50/50
70/30

d) 70
60
50
MgO/PET
850 °C
30/70
50/50
70/30
40 40
30 30
20 20
10 10
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 26. Adsorpcja BR 18 z wodnego roztworu na: a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny: b) Mg(OH)2/PET, c) BMC/PET, d) MgO/PET
Istnieje powiązanie pomiędzy parametrami struktury porowatej (Tabela 8), a przeprowadzonym procesem adsorpcji na materiałach węglowych względem roztworu BR 18 (Rysunek 26, Rysunek 27). W przypadku materiałów zawierających w mieszaninie wyjściowej związek magnezu niestabilnie termicznie, można zauważyć wyraźny wzrost powierzchni i objętości mezoporów wraz ze zwiększaniem jego udziału w mieszaninie wyjściowej. Zwiększenie udziału MgO w mieszanie wyjściowej oraz wzrost temperatury karbonizacji nie wpłynął tak znacząco na rozwój parametrów struktury porowatej zarówno w obszarze mikro-, jak i mezoporów otrzymanych materiałów (Tabela 8). Dlatego też, mezoporowatość materiałów węglowych jest tu
kluczowym czynnikiem wpływającym na przebieg procesu adsorpcji względem
69
roztworu barwnika. Analogiczną tendencję można zaobserwować w przypadku materiałów otrzymanych w zakresie temperatur od 550 °C do 850 °C. Materiały węglowe karbonizowane w 550 °C charakteryzujących się stosunkową małą powierzchnią i objętością mezoporów, co przekłada się na mniejszy stopień usunięcia
zanieczyszczenia (Rysunek 27).
a) 70
60
50
Mg(OH)2/PET
70/30
550
700
850
b) 70
60
50
BMC/PET
50/50

550
700
850
40 40
30 30
20 20
10 10
0
0 10 20 30 40 50
Dawka materiału węglowego[mg]
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]

c) 70
60
MgO/PET
50/50
550
700
850
50
40
30
20
10
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 27. Pomiar adsorpcji BR 18 z wodnego roztworu na węglach aktywnych otrzymanych w różnych temperaturach z mieszaniny: a) Mg(OH)2/PET, b) BMC/PET, c) MgO/PET
Natomiast w przypadku materiałów otrzymanych z mieszaniny BMC/PET oraz
Mg(OH)2/PET zauważa się zwiększenie adsorpcji roztworu BR 18 wraz ze wzrostem
70
temperatury karbonizacji mieszaniny wyjściowej (Rysunek 27 a, b). Tendencja ta wynika ze znacznie większego udziału mezoporów w powyższych materiałach (Tabela 8).
Pomimo dużej różnicy w masach cząsteczkowych barwników, zaobserwowano podobną tendencję w usunięciu barwnika RR 198, jak w przypadku BR 18. Materiały węglowe otrzymane z mieszaniny MgO/PET usuwają RR 198 najmniej efektywnie. Materiał ten charakteryzuje się dosyć niską objętością i powierzchnią mezoporów (Tabela 8), co przekłada się na słabszą zdolność adsorpcyjną roztworu barwnika. Zdolność adsorpcyjna węgli aktywnych otrzymanych z mieszaniny Mg(OH)2/PET lub BMC/PET względem roztworu barwnika RR 198, ulega wyraźnemu wzrostowi wraz ze
zwiększeniu udziału wagowego związku magnezu w mieszaninie wyjściowej.
71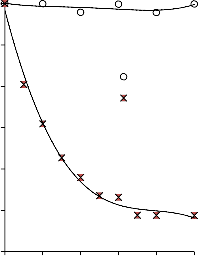
a) 30
25
20
PET CWZ-35

b)
30 Mg(OH)2/PET
850 °C
25
20
30/70
50/50
70/30
15 15
10 10
5 5
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
c) BMC/PET
30 850 °C
25
20
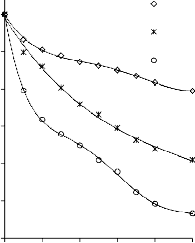
30/70 d)30
50/50
25
70/30
20
MgO/PET
850 °C
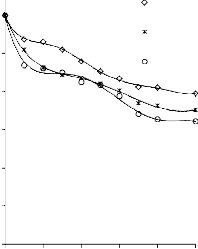
30/70
50/50
70/30
15 15
10 10
5 5
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 28. Adsorpcja RR 198 z wodnego roztworu na: a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny: b) Mg(OH)2/PET, c) BMC/PET, d) MgO/PET
Jak wynika z danych przedstawionych w Tabeli 8, materiały węglowe otrzymane z mieszaniny wyjściowej karbonizowanej w 850 °C, charakteryzowały się największą powierzchnią i objętością mezoporów, co sprzyja adsorpcji adsorbatów o dużej masie cząsteczkowej, jak barwnika RR 198 (Rysunek 28, Rysunek 29).
72
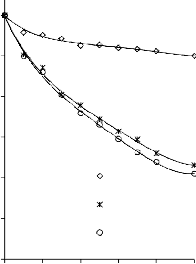
a) b)
30 30
25 25
20 20
15
Mg(OH)2/PET
10 70/30
5
0
550
700
850
15
BMC/PET
10 50/50
5
0
550
700
850
0 10 20 30 40 50
Dawka materiału węglowego [mg]

c)
0 10 20 30 40 50
Dawka materiału węglowego [mg]
30
25
20
15
MgO/PET
10 50/50
5
550
700
850
0
0 10 20 30 40 50
Dawka materiału węglowego [mg]
Rysunek 29. Adsorpcja RR 198 z wodnego roztworu na węglach aktywnych otrzymanych w różnych temperaturach z mieszaniny: a) Mg(OH)2/PET, b) BMC/PET, c) MgO/PET
Najmniejszy udział mezoporów w strukturze badanych adsorbentów wykazały węgle aktywne otrzymane w temperaturze 550 °C, z tego względu wykazują się najmniejszą zdolnością w usuwaniu zanieczyszczenia (RR198) (Rysunek 29).
W przypadku komercyjnego węgla CWZ-35, stopień usunięcia obu zastosowanych roztworów barwnika jest słabszy od materiałów węglowych otrzymanych w 850 °C. Wynika to, z mniejszego udziału mezoporów w strukturze komercyjnego materiału (Tabela 8). Adsorbent w postaci karbonizatu z PET, wykazał nikłą zdolność
adsorpcyjną względem zastosowanych roztworów adsorbatów (Rysunek 28 a).
73
7. Wnioski
1. Zwęglanie poli(tereftalanu etylenu) w mieszaninie z wybranymi związkami magnezu prowadziło do otrzymania bimodalnych (mikro/mezoporowatych) materiałów węglowych.
2. Niezależnie od zastosowanego w mieszaninie wyjściowej z PET niestabilnego termicznie związku magnezu, po zakończonym procesu karbonizacji otrzymany produkt pośredni zawiera materiał węglowy i tlenek magnezu.
3. Woda i ditlenek węgla tworzące się wskutek rozkładu Mg(OH)2 i BMC aktywują in situ karbonizat tworzący się z PET, a tym samym stanowią czynnik porotwórczy, głównie mikroporotwórczy.
4. Tlenek magnezu obecny w karbonizacie sprzyja tworzeniu się mezoporów
w produkcie.
5. Na strukturę porowatą otrzymanych adsorbentów węglowych wpływały przede
wszystkim:
rodzaj zastosowanego związku magnezu w mieszaninie wyjściowej z PET;
udział wagowy związku magnezu w mieszaninie wyjściowej;
temperatura karbonizacji.
6. Znaczny udział mikroporów i mezoporów w strukturze otrzymanych materiałów węglowych, przekłada się na efektywną adsorpcję modelowych zanieczyszczeń wody o szerokim zakresie mas cząsteczkowych.
7. Uzyskiwane materiały węglowe wykazywały częściową okluzję gazowych produktów rozkładu użytych związków magnezu, co może mieć istotny wpływ na poprawność wyznaczenia wydajności procesu.
74
Spis rysunków
Rysunek 1. Model budowy węgla aktywnego ..........................................................................6
Rysunek 2. Proponowany proces technologiczny otrzymywania wysoce porowatych
materiałów .............................................................................................................12
Rysunek 3. Kontrola struktury porowatej materiałó proces aktywacji węgla
formowanego z PET w polimerowych metodą „blending” ...................................15
Rysunek 4. Widmo absorpcyjne UV-Vis: a) fenolu, b) czerwieni anilinowej (BR 18), c)
czerwieni reaktywnej (RR 198) .............................................................................29
Rysunek 5. Schemat otrzymywania materiału węglowego .....................................................33
Rysunek 6. Termogram dla czystego poli(teraftalanu etylenu) ...............................................39
Rysunek 7. Termogram dla czystego Mg(OH)2 oraz mieszaniny wyjściowej
Mg(OH)2/PET (30/70, 50/50, 70/30) ....................................................................40
Rysunek 8. Dyfraktogram XRD przykładowego materiału węglowego otrzymanego
z mieszaniny Mg(OH)2/PET kalcynowanego w temperaturze 550 °C ..................40
Rysunek 9. Termogram dla czystego BMC oraz mieszaniny wyjściowej BMC/PET
(30/70, 50/50, 70/30)..............................................................................................42
Rysunek 10. Dyfraktogramy XRD materiałów węglowych otrzymanych z mieszaniny
BMC/PET 50/50 kalcynowanych w temperaturze 550 °C i 700 °C ...................42
Rysunek 11. Termogram mieszaniny wyjściowej MgO/PET (30/70, 50/50, 70/30)...............43
Rysunek 12. Dyfraktogramy XRD materiału węglowego otrzymanego z mieszaniny
MgO/PET kalcynowanej w temperaturze 850 °C ...............................................44
Rysunek 13. Izotermy adsorpcji/desorpcji N2 w 77 K: a) karbonizatu z PET; b)
komercyjnego węgla CWZ-35.............................................................................45
Rysunek 14. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych
z mieszaniny: a) Mg(OH)2/PET (30,70, 50/50, 70/30) w temperaturze
850 °C; b) Mg(OH)2/PET (70/30) w zakresie temperatur 550 °C – 850 °C .......46
Rysunek 15. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych
z mieszaniny: a) BMC/PET (30,70, 50/50, 70/30) w temperaturze 850 °C;
b) BMC/PET (50/50) w zakresie temperatur 550 °C – 850 °C ...........................46
Rysunek 16. Izotermy adsorpcji/desorpcji N2 w 77 K węgli aktywnych otrzymanych
z mieszaniny: a) MgO/PET (30,70, 50/50, 70/30) w temperaturze 850 °C;
b) MgO/PET (50/50) w zakresie temperatur 550 °C – 850 °C............................47
75
Rysunek 17. Funkcja rozkładu objętości porów wyznaczona metodą BJH dla węgli
aktywnych otrzymanych z mieszaniny: a) BMC/PET (30/70, 50/50, 70/30)
850 °C, b) BMC/PET 50/50 (550 °C – 850 °C), c) Mg(OH)2/PET (30/70,
50/50, 70/30) 850 °C), 70/30 (550 °C), d) MgO/PET 30/70 (700 °C), 50/50
(700 °C, 850 °C) ..................................................................................................51
Rysunek 18. Krzywe TG PET, BMC oraz mieszaniny BMC/PET 50/50 ...............................54
Rysunek 19. Analiza TPD dla materiału węglowego otrzymanego z mieszaniny
BMC/PET 50/50 700 °C......................................................................................55
Rysunek 20. Analiza TPD dla materiałów węglowych otrzymanych z mieszanin:
a) Mg(OH)2/PET 30/70, b) Mg(OH)2/PET 70/30 700 °C ...................................56
Rysunek 21. Analiza TPD dla materiału węglowego otrzymanego z mieszaniny
MgO/PET 50/50 po pirolizie w 700 °C ...............................................................56
Rysunek 22. Proponowany mechanizm tworzenia porów w materiale węglowym ................58
Rysunek 23. Dekonwolucja pasma C (1s) w widmie XPS dla materiałów węglowych
otrzymanych z mieszaniny: a) BMC/PET, b) MgO/PET ....................................60
Rysunek 24. Adsorpcja fenolu z wodnego roztworu na a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny b) Mg(OH)2/PET, c) BMC/PET,
d) MgO/PET ........................................................................................................66
Rysunek 25. Adsorpcja fenolu z wodnego roztworu na węglach aktywnych otrzymanych w różnych temperaturach mieszaniny a) Mg(OH)2/PET,
b) BMC/PET, c) MgO/PET .................................................................................67
Rysunek 26. Adsorpcja BR 18 z wodnego roztworu na: a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny: b) Mg(OH)2/PET, c) BMC/PET,
d) MgO/PET ........................................................................................................69
Rysunek 27. Pomiar adsorpcji BR 18 z wodnego roztworu na węglach aktywnych otrzymanych w różnych temperaturach z mieszaniny: a) Mg(OH)2/PET, b) BMC/PET, c) MgO/PET .....................................................................................70
Rysunek 28. Adsorpcja RR 198 z wodnego roztworu na: a) komercyjnym węglu aktywnym CWZ-35, karbonizacie z PET; węglach aktywnych otrzymanych w 850 °C z mieszaniny: b) Mg(OH)2/PET, c) BMC/PET,
d) MgO/PET ........................................................................................................72
76
Rysunek 29. Adsorpcja RR 198 z wodnego roztworu na węglach aktywnych otrzymanych w różnych temperaturach z mieszaniny: a) Mg(OH)2/PET,
b) BMC/PET, c) MgO/PET .................................................................................73
77
Spis tabel
Tabela 1. Wpływ prekursorów tlenku magnezu na rozwój struktury porowatej
materiałów węglowych ............................................................................................16
Tabela 2. Ponowne wykorzystanie prekursora MgO ...............................................................17
Tabela 3. Zastosowane związki magnezu ................................................................................30
Tabela 4. Modelowe zanieczyszczenia organiczne .................................................................30
Tabela 5. Jakościowy i ilościowy skład mieszanin wyjściowych stosowanych
w badaniach ..............................................................................................................31
Tabela 6. Równania krzywych kalibracji wyznaczonych dla poszczególnych
adsorbatów ...............................................................................................................37
Tabela 7. Wykaz materiałów poddanych analizie termograwimetrycznej ..............................38
Tabela 8. Parametry strukturalne materiałów węglowych wyznaczone na podstawie
niskotemperaturowych izoterm adsorpcji/desorpcji azotu .......................................48
Tabela 9. Średnia wielkość krystalitów MgO materiałów węglowych otrzymanych
z mieszaniny BMC/PET i MgO/PET karbonizowanych w różnym zakresie
temperatur.................................................................................................................52
Tabela 10. Dekonwolucja pasma C (1s) w widmie XPS dla materiałów węglowych
otrzymanych z mieszaniny BMC/PET, MgO/PET ................................................59
Tabela 11. Wydajność węglowa materiałów otrzymanych w wyniku ogrzewania
mieszaniny PET z różnymi związkami magnezu ..................................................63
78
Streszczenie
Głównym celem niniejszej rozprawy doktorskiej było opracowanie jednoetapowej metody otrzymywania porowatych materiałów węglowych bimodalnych, z pominięciem klasycznego etapu aktywacji fizycznej. Jako surowce stosowano mieszaniny poli(tereftalan etylenu) (prekursor węgla) z różnymi związkami magnezu, ulegającymi rozkładowi termicznemu z wydzieleniem produktów gazowych będących typowymi czynnikami aktywującymi (CO2, para wodna). Tlenek magnezu zawarty w produkcie odmywano roztworem kwasu. Stałą pozostałość stanowiły wysoce
porowate materiały węglowe o powierzchni właściwej do 2000 m2/g charakteryzujące
się znaczną powierzchnią zarówno mikroporów jak i mezoporów. Wykazano, że struktura porowata otrzymanych materiałów zależy od udziału wagowego związku magnezu w mieszaninie wyjściowej, jego rodzaju i od temperatury karbonizacji. Określono wpływ produktów rozkładu użytych związków magnezu (stałego – tlenek magnezu i gazowych – CO2/H2O) na rozwój porowatości otrzymanych węgli aktywnych. Zaproponowano również mechanizm tworzenia porów w badanych materiałach węglowych. Dowiedziono, że odpowiedni dobór jakościowy i ilościowy reagentów oraz temperatury procesu otrzymywania stwarza możliwość kontrolowania struktury porowatej otrzymywanych adsorbentów.
Dowiedziono, że uzyskiwane materiały węglowe wykazywały częściową okluzję gazowych produktów rozkładu użytych związków magnezu i potwierdzono wpływ tego zjawiska na poprawność wyznaczenia wydajności procesu.
W pracy zawarto wyniki badań adsorpcji modelowych związków organicznych (fenol, czerwień anilinowa, czerwień reaktywna) o zróżnicowanych masach cząsteczkowych na otrzymanych materiałach węglowych. Potwierdzono, że znaczny udział mikroporów i mezoporów w strukturze otrzymanych materiałów węglowych, przekłada się na efektywną adsorpcję modelowych zanieczyszczeń wody o szerokim zakresie mas
cząsteczkowych.
79
Abstract
The main aim of the presented PhD dissertation was the preparation of bimodal porous carbons in a one-step process through heating of suitable materials in inert atmosphere but without physical activation. The raw materials used were (poly(ethylene terephthalate) (carbon precursor) in a mixture with suitable magnesium compounds. The latter were thermally unstable and decomposed during heating releasing gases (CO2 and/or H2O) normally used as activating agents. Magnesium oxide contained in the product left after heating was washed out using acid solution. Remained carbon residues were highly porous carbons revealing high specific surface area (up to ~2000 m2/g) with considerable contribution of micropores and mesopores. It was proved that the porous structure of the carbons strongly depended from the qualitative and quantitative composition of the magnesium compounds/PET mixtures and as well as the temperature of carbonization process. Effect of MgO and CO2/H2O released in situ on the development of porosity is studied. On the basis of the obtained results the pore creation mechanism in the magnesium compound/PET system was proposed. It is proved that
pore structure of the prepared materials can be controlled to some extent by varying preparation conditions and composition of the raw mixtures. As confirmed by additional experiments, partial occlusion of the gaseous products of the magnesium compounds decomposition process, may cause to improper estimation of carbon yield. The thesis contains results confirming suitability of the porous carbons for adsorption of model contaminants (phenol, Basic Red 18, and Reactive Red 198). As found significant, microporosity and mesoporosity of the studied materials makes them suitable for
adsorption of adsorbates of wide range molecular weights.
80
Wykaz literatury cytowanej
1 B. H. Hameed, A. A. Rahman, Removal of phenol from aqueous solutions by adsorption onto activated carbon prepared from biomass material, Journal of Hazardous Materials 160 (2008) 576 – 581
2 M. A. Lillo-Ródenas, A. J. Fletcher, K. M. Thomas, D. Cazorla-Amorós, A. Linares-
Solano, Competitive adsorption of a benzene-toluene mixture on activated carbons at low concentration, Carbon 44 (2006) 1455 – 1463
3 S. Sumathi, S. Bhatia, K. T. Lee, A. R. Mohamed, Adsorption isotherm models and
properties of SO2 and NO removal by palm shell activated carbon supported with cerium (Ce/PSAC), Chemical Engineering Journal 162 (2010) 194 – 200
4 Q. Wen, C. Li, Z. Cai, W. Zhang, H. Gao, L. Chen, G. Zeng, X. Shu, Y. Zhao, Study
on activated carbon derived from sewage sludge for adsorption of gaseous formaldehyde, Bioresource Technology 102 (2011) 942 – 947
5 M. P. Cal, M. J. Rood, S. M. Larson, Removal of VOCs from humidified gas streams
using activated carbon cloth, Gas Separation & Purification 10 (1996) 117 – 121
6 T. Nakai, S. N. Kartal, T. Hata, Y. Imamura, Chemical characterization of pyrolysis liquids of w ood-based composites and evaluation of their bio-efficiency, Building and Environment 42 (2007) 1236 – 1241
7 H. Benaddi, D. Legras, J. N. Rouzaud, F. Beguin, Influence of the atmosphere in the
chemical activation of wood by phosphoric acid, Carbon 36 (1998) 306 – 309
8 M. Jasieńko-Hałat, K. Kędzior, Comparison of molecular sieve properties in microporous chars from low-rank bituminous coal activated by steam and carbon dioxide, Carbon 43 (2005) 944 – 953
9 L. Ballice, R. Reimert, Temperature-programmed co-pyrolysis of Turkish lignite with
polypropylene, Journal of Analytical and Applied Pyrolysis 65 (2002) 207 – 219
10 A. Aygun, S. Yenisoy-Karakasb, I. Duman, Production of granular activated carbon from fruit stones and nutshells and evaluation of their physical, chemical and adsorption properties, Microporous and Mesoporous Materials 66 (2003) 189 – 195
11 W. T. Tsai, M. K. Lee, Y. M. Chang, Fast pyrolysis of rice straw, sugarcane bagasse
and coconut shell in an induction-heating reactor, Journal of Analytical and Applied
Pyrolysis 76 (2006) 230 – 237
81
12 F. Lian, B. Xing, L. Zhu, Comparative study on composition, structure, and adsorption behavior of activated carbons derived from different synthetic waste polymers, Journal of Colloid and Interface Science 360 (2011) 725 – 730
13 Q. Wang, X. Liang, W. Qiao, C. Liu, X. Liu, L. Zhan, L. Ling, Preparation of
polystyrene-based activated carbon spheres with high surface area and their adsorption to dibenzothiophene, Fuel Processing Technology 90 (2009) 381 – 387
14 R. C. Bansal, M. Goyal, Adsorpcja na węglu aktywnym, Wydawnictwo Naukowo-
Techniczne, Warszawa (2009)
15 J. Nawrocki, Uzdatnianie wody: procesy fizyczne i biologiczne, Wydawnictwo
Naukowe PWN, Warszawa (2010)
16 H. Marsh, F. Rodriguez-Reinoso, Activated Carbon, Elsevier Science & Technology
Books, Oxford (2006)
17 H. Jasińska, A. Świątkowski, J. Choma, Węgiel aktywny, Wydawnictwo Naukowo- Techniczne, Warszawa (1985)
18 D. M. Mackay, P. V. Roberts, The influence of pyrolysis conditions on the
subsequent gasification of lignocellulosic chars, Carbon 20 (1982) 105 – 111
19 W. M. A. W. Daud, W. S. W. Ali, M. Z. Sulaiman, The effects of carbonization temperature on pore development in palm-shell-based activated carbon, Carbon 38 (2000) 1925 – 1932
20 A. Marcilla, M. Asensio, I. Martin-Gullón, Influence of the carbonization heating rate
on the physical properties of activated carbons from a sub-bituminous coal, Carbon 34 (1996) 449 – 456
21 A. C. Lua, T. Yand, J. Gou, Effects of pyrolysis conditions on the properties of
activated carbons prepared from pistachio-nut shells, Journal of Analytical and Applied
Pyrolysis 72 (2004) 279 – 287
22 G. San Miguel, G. D. Fowler, C. J. Sollars, A study of the characteristics of activated carbons produced by steam and carbon dioxide activation of waste tyre rubber, Carbon
41 (2003) 1009 – 1016
23 G. San Miguel, G. D. Fowler, C. J. Sollars, A study of the characteristics of activated carbons produced by steam and carbon dioxide activation of waste tyre rubber, Carbon
41 (2003) 1009 – 1016
82
24 C. Bouchelta, M. S. Medjram, O. Bertrand, J. P. Bellat, Preparation and characterization of activated carbon from date Stones by physical activation with steam, Journal of Analytical and Applied Pyrolysis 82 (2008) 70 – 77
25 M. L. Sekirifa, M. Hadj-Mahammed, S. Pallier, L. Baameur, D. Richard, A. H. Al.-
Dujaili, Preparation and characterization of an activated carbon from a date Stones vartiety by physical activation with carbon dioxide, Journal of Analytical and Applied Pyrolysis 99 (2013) 155 – 160
26 M. Paderewski, Adsorpcja i adsorbenty, Politechnika Szczecińska, Szczecin (1982)
27 Ö. Şahin, C. Saka, Preparation and characterization of activated carbon from acorn shell by physical activation with H2O–CO2 in two-step pretreatment, Bioresource Technology 136 (2013) 163 – 168
28 M. Ahmedna, W .E. Marshall, R. M. Rao, Surface properties of granular activated
carbons from agricultural by-products and their effects on raw sugar decolorization, Bioresource Technology 71 (2000) 103 – 112
29 S. Guo, J. Peng, W. Li, K. Yang, L. Zhang, S. Zhang, H. Xia, Effects of CO2
activation on porous structures of coconut shell-based activated carbons, Applied
Surface Science 255 (2009) 8443 – 8449
30 K. Fu, Q. Yue, B. Gao, Y. Sun, L. Zhu, Preparation, characterization and application of lignin-based activated carbon from black liquor lignin by steam activation, Chemical Engineering Journal 228 (2013) 1074 – 1082
31 M. Molina-Sabio, M. Gonzalez, F. Rodriguez-Reinoso, A. Sepulveda-Escribano,
Effect of steam and carbon dioxide activation in the micropore size distribution of activated carbon, Carbon 34 (1996) 505 – 509
32 K. Laszlo, E. Tombacz, K. Josepovits, Effect of activation on the surface chemistry of
carbons from polymer precursors, Carbon 39 (2001) 1217 – 1228
33 M. T. Izquierdo, A. Martínez de Yuso, B. Rubio, M. R. Pino, Conversion of almond shell to activated carbons: Methodical study of the chemical activation based on an experimental design and relationship with their characteristics, Biomass and Bioenergy
35 (2011) 1235 – 1244
34 J. Hayashia, A. Kazehayaa, K. Muroyamaa, A. P. Watkinson, Preparation of activated carbon from lignin by chemical activation, Carbon 38 (2000) 1873 – 1878
35 J. Hayashia, A. Kazehayaa, K. Muroyamaa, A. P. Watkinson, Preparation of activated
carbon from lignin by chemical activation, Carbon 38 (2000) 1873 – 1878
83
36 N. Yalçın, V. Sevinç, Studies of the surface area and porosity of activated carbons
prepared from rice husks, Carbon 38 (2000) 1943 – 1945
37 A. H. Basta, V. Fierro, H. El-Saied, A. Celzard, 2-Steps KOH activation of rice straw: An efficient method for preparing high-performance activated carbons, Bioresource Technology 100 (2009) 3941 – 3947
38 R. L. Tseng, Physical and chemical properties and adsorption type of activated carbon
prepared from plum kernels by NaOH activation, Journal of Hazardous Materials 147 (2007) 1020 – 1027
39 L. Y. Hsu, H. Teng, Influence of different chemical reagents on the preparation of
activated carbons from bituminous coal, Fuel Processing Technology 64 (2000) 155 –
166
40 A. Heidari, H. Younesi, A. Rashidi, A. A. Ghoreyshi, Adsorptive removal of CO2 on highly microporous activated carbons prepared from Eucalyptus camaldulensis wood: Effect of chemical activation, Journal of the Taiwan Insitute of Chemical Engineers http://dx.doi.org/10.1016/j.jtice.2013.06.007 (2013)
41 F. Caturla, M. Molina-Sabio, F. Rodrıguez-Reinoso, Preparation of activated carbon
by chemical activation with ZnCl2, Carbon 29 (1991) 999 – 1007
42 M. Jagtoyen, M. Thwaites, J. Stencel, B. McEnaney, F. Derbyshire, Adsorbent carbon synthesis from coals by phosphoric acid activation, Carbon 30 (1992) 1089 – 1096
43 K. Kierzak, M. Król, H. Machnikowska, G. Gryglewicz, J. Machnikowski, Aktywacja
alkaliami – możliwości i ograniczenia, Węgiel Aktywny w Ochronie Środowiska
i Przemyśle (2008) 198 – 206
44 F. Lian, B. Xing, L. Zhu, Comparative study on composition, structure, and adsorption behaviour of activated carbons derived from different synthetic waste polymers, Journal of Colloid and Interface Science 360 (2011) 725 – 730
45 J. Choma, M. Kloske, Otrzymywanie i właściwości impregnowanych węgli
aktywnych, Ochrona Środowiska 2 (1999) 1 – 17
46 H. Teng, T. S. Yeh, L. Y. Hsu, Preparation of activated carbon from bituminous coal with phosphoric acid activation, Carbon 36 (1998) 1387 – 1395
47 A. Ahmadpour, D. D. Do, The preparation of active carbons from coal by chemical
and physical activation, Carbon 34 (1996) 471 – 479
84
48 M. Molina-Sabio, F. Rodrıguez-Reinoso, F. Caturla, M. J. Sellés, Development of
porosity in comined phosphoric acid – carbon dioxide activation, Carbon 34 (1996) 457
– 462
49 N. V. Sych, N. T. Tsyba, V. V. Strelko, Effect of combined activation on preparation of high porous active carbons from granulated post-consumer polyethyleneterephthalate, Applied Surface Science 252 (2006) 8062 – 8066
50 M. A. Ahmad, W.M.A. wan Daud, M.K. Aroua, Adsorption kinetics of various gases
in carbon molecular sieves (CMS) produced from palm shell, Colloids and Surfaces A: Physicochemical and Engineering Aspects 312 (2008) 131 – 135
51 W. M. A. wan Daud, M. A. Ahmad, M. K. Aroua, Carbon molecular sieves from
palm shell: Effect of the benzene deposition Times on gas separation properties, Separation and Purification Technology 57 (2007) 289 – 293
52 O. N. Baklanova, G. V. Plaksin, V. A. Drozdov, V. K. Duyakin, N. V. Chesnokov, B.
N. Kuznetsov, Preparation of microporous sorbents from cedar nuthells and hydrolytic lignin, Carbon 41 (2003) 1793 – 1800
53 A. A. Lizzo, M. Rostam-Abadi, Production of carbon molecular sieves from Illinois
coal, Fuel Processing Technology 34 (1993) 97 – 122
54 T. Kyotani, T. Nagai, S. Inoue, A. Tomita, Formation of new type of porous carbon by carbonization in zeolite nanochannels, Chemistry of Materials 9 (1997) 609 – 615
55 P. X. Hou, T. Yamaguchi, H. Orikasa, T. Kyotani, An easy method for the synthesis
of ordered microporous carbons by the template technique, Carbon 43 (2005) 2618 –
2641
56 J. S. Lee, S. H. Joo, R. Ryoo, Synthesis of mesoporous silicas of controlled pore w all thickness and their replication to ordered nanoporous carbons with various pore dimensions, Journal of the American Chemical Society 12 (2002) 1156 – 1157
57 N. Patel, K. Okabe, A. Oya, Designing carbon materials with unique shapes using
polimer blending and coating techniques, Carbon 40 (2002) 315 – 320
58 E. Yasuda, M. Inagaki, K. Kaneko, M. Endo, A. Oya, Y. Tanabe, Carbon Alloys, Novel Concepts to Develop Carbon Science and Technology, Elsevier Science, Oxford (2003)
59 M. Inagaki, H. Miura, H. Konno, A new simple process for carbon coating of ceramic
particles using poly(vinyl chloride), Journal of the European Ceramic Society 18 (1998)
1011 – 1015
85
60 M. Inagaki, S. Kobayashi, F. Kojin, N. Tanaka, T. Morishita, B. Tryba, Pore structure of carbon coated on ceramic particles, Carbon 42 (2004) 3153 – 3158
61 T. Morishita, T. Tsumura, M. Toyoda, J. Przepiórski, A. W. Morawski, H. Konno, M.
Inagaki, A review of the control of pore structure in MgO - templated nanoporous carbons, Carbon 48 (2010) 2690 – 2707
62 T. Morishita, Y. Soneda, T. Tsumura, M. Inagaki, Preparation of porous carbons from
thermoplastic precursors and their performance for electric double layer capacitors, Carbon 44 (2006) 2360 – 2367
64 H. Konno, H. Onishi, N. Yoshizawa, K. Azumi, MgO-templated nitrogen-containing carbons derived from different organic compounds for capacitor electrodes, Journal of Power Sources 195 (2010) 667 – 673
65 A. Esfandiari, T. Kaghazchi, M. Soleimani, Preparation and evolution of activated
carbons by physical activation of polyethyleneterephtalate (PET) wastes, Journal of the
Taiwan Institute of Chemical Engineers 43 (2012) 631 – 637
66 Błędzik A.K., Recykling materiałów polimerowych, Wydawnictwo Naukowo
Techniczne, Warszawa 1997
67 M. P. Stevens, Wprowadzenie do chemii polimerów, Państwowe Wydawnictwa
Naukowe, Warszawa (1983)
68 M. Marzec, B. Tryba, R. J. Kaleńczuk, A.W. Morawski, Poly(ethylene terephthalate)
as a source for activated carbon, Polymer for Advanced Technologies 10 (1999) 588 –
595
69 J. B. Parra, C. O. Ania, A. Arenillas, J. J. Pis, Textural characterization of activated carbons obtained from poly(ethylene terephthalate) by carbon dioxide activation, Studies in Surface Science and Catalysis, 144 (2002) 537 – 543
70 I. Fernández-Morales, M. C. Almazán-Almazán, M. Pérez-Mendoza, M. Domingo-
García, F. J. López-Garzón, PET as precursor of microporous carbons: preparation and
characterization, Microporous and Mesoporous Materials 80 (2005) 107 – 115
71 M. T. Kartel, N. V. Sych, M. M. Tsyba, V. V. Strelko, Preparation of porous carbons by chemical activation of polyethyleneterephthalate, Carbon 44 (2006) 1019 – 1022
72 M. Domingo-García, J. A. Fernández, M. C. Almazán-Almazán, F.J. López-Garzón,
F. Stoeckli, T. A. Centeno, Poly(ethylene terephthalate)-based carbons as electrode material in supercapacitors, Journal of Power Sources 195 (2010) 3810 – 3813
86
73 K. Laszlo, A. Szucs, Surface characterization of polyethyleneterephthalate (PET) based activated carbon and the effect of pH on its adsorption capacity from aqueous phenol and 2,3,4-trichlorophenol solutions, Carbon 39 (2001) 1945 – 1953
74 K. Laszlo, A. Bota, I. Dekany, Effect of heat treatment on synthetic carbon
precursors, Carbon 41 (2003) 1205 – 1214
75 K. Laszlo , E. Tombacz , K. Josepovits, Effect of activation on the surface chemistry of carbons from polymer precursors, Carbon 39 (2001) 1217 – 1228
76 W. Bratek, A. Świątkowski, M. Pakuła, S. Biniak, M. Bystrzejewski, R. Szmigielski,
Characteristics of activated carbon prepared from waste PET by carbon dioxide activation, Journal of Analytical and Applied Pyrolysis 100 (2013) 192 – 198
77 K. Nakagawa, A. Namba, S. R. Mukai, H. Tamon, P. Ariyadejwanich,
W. Tanthapanichakoon, Adsorption of phenol and reactive dye from aqueous solution on activated carbons derived from solid wastes, Water Research 38 (2004) 1791 – 1798
78 J. B. Parra, C. O. Ania, A. Arneillas, F. Rubiera, J. J. Pis, High value carbon materials
from PET recycling, Applied Surface Science 238 (2004) 304 – 308
79 M. T. Kartel, N.V. Sych, M. M. Tsyba, V. V. Strelko, Preparation of porous carbons by chemical activation of polyethyleneterephthalate. Carbon 44 (2006) 1019 – 1022
80 M. Gryta, A. W. Morawski, Karbo-Energochemia-Ekologia 43 (1998) 167-170
81 M. Gryta, A. W. Morawski, III Ogólnopolska Konferencja Naukowa „Inżynieria
Procesowa w Ochronie Środowiska”, Materiały Konferencyjne, Jachranka (1997) 306 –
310
82 M. C. Almazán-Almazán, M. Pérez-Mendoza, M. Domingo-García, I. Fernández- Morales, F. Javier López, F. Javier López-Garzón, The influence of the process conditions on the characteristics of activated carbons obtained from PET de- polymerisation, Fuel Processing Technology 91 (2010) 236 – 242
83 M. C. Almazán-Almazán, M. Pérez-Mendoza, F. J. López-Domingo, I. Fernández-
Morales, M. Domingo-García, F. J. López-Garzón, A new method to obtain microporous carbons from PET: Characterization by adsorption and molecular simulation, Microporous and Mesoporous Materials 106 (2007) 219 – 228
84 A. L. Kowal, Oczyszczanie wody : podstawy teoretyczne i technologiczne, Procesy i
Urządzenia, Wydawnictwo Naukowe PWN, Warszawa (2007)
85 B. C. Moore, F. S. Cannon, J. A. Westrick, D. H. Metz, C. A. Shrive, J. DeMarco, D. J. Hartman, Changes in GAC pore structure during full-scale water treatment at
87
Cincinnati: a comparison between virgin and thermally reactivated GAC, Carbon 39 (2001) 789 – 807
86 H. P. Boehm, Some aspects of the surface chemistry of carbon blacks and other
carbons, Carbon 32 (1994) 759 – 769
87 G. Newcombe, M. Drikas, Adsorption of NOM onto activated carbon: electrostatic and non-electrostatic effects, Carbon 35 (1997) 1239 – 1250
88 P. Pendleton, S. H. Wong, R. Schuman, G. Levay, R. Denoyel, J. Rouquerol,
Properties of activated carbon controlling 2 methylisoborneol adsorption, Carbon 35 (1997) 1141 – 1149
89 G. Newcombe, Activated carbon and soluble humic substances: adsorption,
desorption, and surface charge effects, Journal of Colloid and Interface Science 164 (1994) 452 – 462
90 C. Faur-Brasquet, K. Kadirvelu, P. Le Cloirec, Removal of metal ions from aqueous
solution by adsorption onto activated carbon sloths: adsorption competition with organic matter, Carbon 40 (2002) 2387 – 2392
91 R. D. Vidic, M. T. Suidan, U. K. Traegner, G. F. Nakhla, Adsorption isotherms:
illusive capacity and role of oxygen, Water Research 24 (1990) 1187–1195
92 P. A. M. Mourão, P. J. M. Carrott, M. M. L. Ribeiro Carrott, Application of different equations to adsorption isotherms of phenolic compounds on activated carbons prepared from cork, Carbon 44 (2006) 2422 – 2429
93 L. Lei, P. A. Quinlivan, D. R.U. Knappe, Effects of activated carbon surface
chemistry and pore structure on the adsorption of organic contaminants from aqueous solution, Carbon 40 (2002) 2085 – 2100
94 R. L. Tseng, F. C. Wu, R. S. Juang, Liquid-phase adsorption of dyes and phenols
using pinewood-based activated carbons, Carbon 41 (2003) 487 – 495
95 M. O. Corapcioglu, C. P. Huang, The adsorption of heavy metals onto hydrous activated carbon, Water Research 21 (1987) 1031 – 1044
96 V. C. Srivastava, I. D. Mall, I. M. Mishra, Adsorption of toxic metal ions onto
activated carbon: Study of sorption behaviour characterization and kinetics, Chemical
Engineering and Processing: Process Intensification 48 (2008) 1269 – 1280
97 M. Kobyam E. Demirbas, E. Sentruk, M. Ince, Adsorption of heavy metal ions from aqueous solutions by activated carbon prepared from apricot stone, Bioresource Technology 96 (2005) 1518 – 1521
88
98 G. Cimino, A. Passerini, G. Toscano, Removal of toxic cations and Cr(VI) from aqueous solution by hazelnut shell, Water Research 34 (2000) 2955 – 2962
99 H. Petrov, T. Budinova, I. Khovesov, Adsorption of zinc, cadmium, copper and lead
ions on oxidised anthracite, Carbon 30 (1992) 135-139
100 K. Mohanty, D. Das, M. N. Biswas, Adsorption of phenol from aqueous solutions using activated carbons prepared from Tectona grandis sawdust by ZnCl2 activation, Chemical Engineering Journal 115 (2005) 121 – 131
101 A. Dąbrowski, P. Podkościelny, Z. Hubicki, M. Barczak, Adsorption of phenolic
compounds by activated carbon—a critical review, Chemosphere 58 (2005) 1049 –
1070
102 Md. Zahangir Alam, Emad S. Ameem, Suleyman A. Muyibi, Nassereldeen A. Kabbashi, The factors affecting the performance of activated carbon prepared from oil palm empty fruit bunches for adsorption of phenol, Chemical Engineering Journal 155 (2009) 191 – 198
103 K. H. Park, M. S. Balathanigaimani,W. G. Shim, J. W. Lee, H. Moon, Adsorption
characteristics of phenol on novel corn grain-based activated carbons, Microporous and
Mesoporous Materials 127 (2010) 1 – 8
104 S. I. Kim, T. Yamamoto, A. Endo, T. Ohmori, M. Nakaiwa, Adsorption of phenol and reactive dyes from aqueous solution on carbon cryogel microspheres with controlled porous structure, Microporous and Mesoporous Materials 96 (2006) 191 –
196
105 B. H. Hameed, A. A. Rahman, Removal of phenol from aqueous solutions by adsorption onto activated carbon prepared from biomass material, Journal of Hazardous Materials 160 (2008) 576 – 581
106 M. Kilic, E. Apaydin-Varol, A. E. Pütün, Adsorptive removal of phenol from
aqueous solutions on activated carbon prepared from tobacco residues: Equilibrium, kinetics and thermodynamics, Journal of Hazardous Materials 189 (2011) 397 – 403
107 I. I. Salame, T. J. Bandosz, Role of surface chemistry in adsorption of phenol on
activated carbons, Journal of Colloid and Interface Science 264 (2003) 307 – 312
108 R. C. Bansal, D. Aggarwal, M. Goyal, B. C. Kaistha, Influence of carbon-oxygen surface gropus on the adsorption of phenol by activated carbons 9 (2002) 290 – 296
89
109 J. L. Figueiredo, N. Mahata, M. F. R. Pereira, M. J. Sánchez Montero, F. Salvador, Adsorption of phenol on supercritically activated carbon fibres: Effect of texture and surface chemistry, Journal of Colloid and Interface Science 357 (2011) 210 – 214
110 A. Lisiak, M. Miklas, Problemy szkodliwości barwników azowych, Przegląd
włókienniczy, Włókno. Odzież. Skóra 3 (2007) 38
111 G. Mezohegyi, F. P. van der Zee, J. Font, A. Fortuny, A. Fabregat, Towards advanced aqueous dye removal processes: A short review on the versatile role of activated carbon, Journal of Environmental Management 102 (2012) 148 – 164
112 J. J. M. Órfão, A. I. M. Silva, J. C. V. Pereira, S. A. Barata, I. M. Fonseca, P. C. C.
Faria, M. F. R. Pereira, Adsorption of a reactive dye on chemically modified activated carbon – Influence of pH, Journal of Colloid and Interface Science 296 (2006) 480 –
489
113 E. N. El Qada, S.J. Allen, G.M. Walker, Adsorption of basic dyes from aqueous solution onto activated carbons, Chemical Engineering Journal 135 (2008) 174 – 184
114 H. Tamai, T. Yoshida, M. Sasaki, H. Yasuda, Dye adsorption on mesoporous
activated carbon fiber obtained from pitch containing yttrium complex, Carbon 37 (1999) 983 – 989
115 E. Lorence – Grabowska, G. Gryglewicz, Adsorption characteristics of Congo Red
on coal-based mesoporous activated carbon, Dyes and Pigments 74 (2007) 34 – 40
116 C. Pelekani, V. L. Snoeyink, Competitive adsorption in natural water: role of activated carbon pore size, Water Research 33 (1999) 1209 – 1219
117 E. Lorenc-Grabowska, G. Gryglewicz, Adsorption of lignite-derived humic acids on
coal-based mesoporous activated carbons, Journal of Colloid and Interface Science 284 (2005) 416 – 423
118 J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe, D. R. Bloch, Polymer
Handbook, John Wiley & Sons, New York (2005)
119 S. Lowell, J.E. Shields, Powder Surface Area and Porosity, Chapman and Hall, London (1991)
120 S. J. Gregg, K.S.W. Sing: Adsorption, Surface Area and Porosity. Academic Press,
New York (1982)
121 M.M. Dubinin, Water vapour adsorption and the micoporous structure of carbonaceous adsorbent. Carbon 18 (1980) 355 – 364
90
122 E.P. Barrett, L.G. Joyner, P.P. Halenda: The determination of pore volume and area distribution in porous substances. I. Computations from nitrogen isotherms. Journal of the American Chemical Society 73 (1951) 373 – 380
123 J. F. Moulder, W. F. Stickle, P. E. Sobol, K. D. Bomben, Handbook of X-ray
Photoelectron Spectroscopy, Physical Electronic, USA (1995)
124 J. Przepiórski, J. Karolczyk, K. Takeda, T. Tsumura, M. Toyoda, A.W. Morawski, Porous carbon obtained by carbonization of PET mixed with basic magnesium carbonate: pore structure and pore creation mechanism, Industrial & Engineering Chemistry Research 48 (2009) 7110 – 7116
125 L. Yan, J. Zhuang, X. Sun, Z. Deng, Y. Li, Formation of rod-like Mg(OH)2
nanocrystallites under hydrothermal conditions and the conversion to MgO nanorods by thermal dehydration, Materials Chemistry and Physics 76 (2002) 119 – 122
126 G. Helou, S.A. Tariq, The Pyrolysis of Basic Magnesium Carbonate Trihydrate.
Thermochimica Acta 228 (1993) 123 – 126
127 K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, R.A. Pierotti, J. Rouquerol, T. Siemieniawska: Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure and Applied Chemistry
57 (1985) 603 – 619
128 F. Rodriguez-Reinoso, M. Molina-Sabio, Textural and Chemical Characterization of
Microporous Carbons, Adances in Colloid Interface Science 271 (1998) 76 – 77
129 G. S. Szymański, Z. Karpiński, S. Biniak, A. Świątkowski, The Effect of the Gradual Thermal Decomposition of Surface Oxygen Species on the Chemical and Catalytic Properties of Oxidized Activated Carbon, Carbon 40 (2002) 2627 – 2639
130 M. E. Sanchez, A. Moran, A. Escapa, L. F. Calvo, O. Martinez, Simultaneous Thermogravimetric and mass spectrometric analysis of the pyrolysis of municipal solid wastes and polyethylene terephthalate, Journal of Thermal Analysis and Calorimetry 90 (2007) 209 – 215
131 O. Terakado, M. Hirasawa, Effect of metal oxides on the pyrolysis residues of
poly(ethylene terephthalate): formation of carbonaceous submicron, nano-scale filaments and mesoporous compounds, Journal of Analytical and Applied Pyrolysis 73 (2005) 248 – 256
91