
Praca magisterskaWpływ dodatku wybranych substancji nieorganicznych na właściwości węgli aktywnych otrzymywanych z politereftalanu etylenu (PET) - fragment
Spis treści
1. Węgiel aktywny
1.1 Struktura węgla aktywnego
1.2 Otrzymywanie węgli aktywnych
1.2.1 Surowce do otrzymywania węgli aktywnych
1.2.2 Karbonizacja
1.2.3 Aktywacja
1.3 Kontrola struktury porowatej
1.3.1 Kontrola struktury mikroporowatej
1.3.2 Kontrola struktury mezoporowatej
1.4 Poli(tereftalan etylenu) jako prekursor węgli aktywnych
2. Cel i zakres pracy
3. Charakterystyka surowców i odczynników
3.1 Poli(tereftalan etylenu) - (PET)
3.2 Związki magnezu
3.3 Charakterystyka odczynników stosowanych w pomiarach
4. Preparatyka węgli aktywnych
4.1 Przygotowanie mieszaniny PET ze związkami magnezu
4.2 Karbonizacja próbek
4.3 Przemywanie próbek 10-procentowym roztworem kwasu solnego
5. Metody analityczne
5.1 Pomiar termograwimetryczny (TG)
5.2 Pomiar izoterm adsorpcji, desorpcji azotu w temperaturze 77K
5.3 Analiza składu fazowego otrzymanych węgli aktywnych metodą dyfrakcji rentgenowskiej (XRD)
5.4 Transmisyjna mikroskopia elektronowa (TEM)
5.5 Pomiar adsorpcji czerwieni reaktywnej z roztworu wodnego na otrzymanych węglach aktywnych metodą spektrofotometrii UV-VIS
5.6 Pomiar adsorpcji fenolu z roztworu wodnego na otrzymanych węglach aktywnych metodą spektrofotometrii UV-VIS
5.7 Pomiar adsorcpji kwasów humusowych z roztworu wodnego na otrzymanych węglach aktywnych
5.7.1 Pomiar zawartości ogólnego węgla organicznego (TOC)
5.7.2 Metoda spektrofotometrii UV-VIS
6. Omówienie wyników i dyskusja
6.1 Analiza XRD
6.2 Transmisyjna mikroskopia elektronowa
6.3 Wyniki pomiarów termograwimetrycznych wybranych próbek
6.4 Pomiar izoterm adsorpcji, desorpcji azotu w temperaturze 77K
7. Próby oszacowania przydatności otrzymanych materiałów węglowych do procesów adsorpcji
7.1 Pomiar adsorpcji fenolu z wodnego roztworu na otrzymanych węglach aktywnych
7.2 Pomiar adsorpcji czerwieni reaktywnej z wodnego roztworu na otrzymanych węglach aktywnych
7.3 Pomiar adsorpcji kwasów humusowych z roztworu na otrzymanych węglach aktywnych
7.3.1 Metoda spektrofotometrii UV-VIS
7.3.2 Pomiar stężenia kwasu humusowego na podstawie pomiaru zawartości ogólnego węgla organicznego
8. Wnioski
Bibliografia
Spis rysunków
Spis tabel
1. Węgiel aktywny
XXI wiek stoi pod znakiem konsumpcji, szybko rozwijającego się przemysłu oraz gospodarki, wiąże się to z silnymi zanieczyszczeniami środowiska. Zanieczyszczenia te w dużej mierze związane są z coraz szerzej stosowanymi w przemyśle artykułami produkowanymi z polimerów. Znaczną ilość odpadów stanowią polimery jak np. poli(tereftalanetylenu), z którego produkowane są butelki oraz inne opakowania. Alternatywą dla recyklingu materiałów polimerowych (wysokie koszty, wymagana duża czystość materiału) jest wykorzystanie ich do produkcji adsorbentów węglowych., które mogą posłużyć do oczyszczania środowiska (woda, powietrze). Ważnym przykładem jest stosowanie do tego celu odpadowego poli(tereftalanu etylenu), materiału bardzo trudno ulegającego naturalnemu rozkładowi, a jednocześnie charakteryzującym się wysoką zawartością węgla (62,5%). Z tworzywa tego podejmowane są próby otrzymywania wyżej wspomnianych adsorbentów węglowych [1].
Węgiel aktywny jest adsorbentem, którego prapoczątki zastosowania wiążą się z medycyną. Hipokrates i jego uczniowie zalecali zasypywanie węglem drzewnym ran w celu usuwania ich przykrego zapachu. Węgiel aktywny znalazł też zastosowanie w przemyśle cukierniczym jako środek do odbarwiania syropów cukrowych. W XVI wieku odkryto, że węgiel drzewny jest zdolny do pochłaniania niektórych gazów oraz do adsorpcji z fazy ciekłej. [2]
Węgle aktywne należą do grupy materiałów węglowych o bardzo silnie rozwiniętej porowatości i powierzchni właściwej. W ciągu ostatnich dwudziestu lat obserwuje się gwałtowny rozwój zapotrzebowania na te materiały, a towarzyszy temu nie mniej dynamiczny rozwój nauki o nich samych, jak i o zjawisku adsorpcji na ich powierzchni. Węgle aktywne znajdują coraz szersze zastosowanie głównie jako adsorbenty i nośniki katalizatorów, a ostatnio jako materiały elektrodowe w chemicznych źródłach prądu. Rosnące zagrożenie środowiska naturalnego człowieka powodowane przez przemysłowe stwarza dla węgli aktywnych nowe perspektywy zastosowania o coraz większym znaczeniu[2].
Do procesów o największym znaczeniu przemysłowym i użytkowym z zastosowaniem adsorbentów węglowych zalicza się: usuwanie siarkowodoru z gazu ziemnego, oczyszczanie wody pitnej (usuwanie zanieczyszczeń organicznych) i odpadowej (pestycydy, detergenty, metale ciężkie, barwniki), odzyskiwanie par węglowodorów w trakcie przeładunku rozpuszczalników, usuwanie par rtęci z powietrza, oczyszczanie powietrza w systemach klimatyzacyjnych. W mniejszej skali wykorzystywane jako porowate materiały w postaci sit molekularnych do rozdziału mieszanin gazowych oraz jako katalizatory i nośniki katalizatorów. Szacuje się, że światowa produkcja różnego typu węgli aktywnych wynosi obecnie około 390 000 ton na rok i wzrasta w tempie około 7% rocznie. Do krajów o największym zużyciu węgli aktywnych zalicza się Japonię i Stany Zjednoczone. Szybki wzrost znaczenia węgli aktywnych w tych krajach jest spowodowany zarówno zwiększonym potrzebami w wymienionych tradycyjnych dziedzinach zastosowań jak i rozwojem nowych technologii opartych na wykorzystaniu materiałów porowatych. Należą do nich m.in. nowoczesne systemy magazynowania energii [3].
1.1 Struktura węgla aktywnego
Węgiel aktywny jest materiałem o charakterze hydrofobowym. Węgiel aktywny to substancja składająca się z węgla pierwiastkowego w formie bezpostaciowej (sadza) i drobnokrystalicznego grafitu. Charakteryzuje się dużą powierzchnią właściwą (jednostka powierzchni na jednostkę masy 500 – 2500 m2/g). Charakterystyczny dla tego materiału jest nierównomierny rozkład atomów węgla oraz obecność licznych mikropęknięć i szczelin (porów). Na strukturę porowatą składają się połączone ze sobą pory o różnym kształcie i wielkości.
Struktura węgla aktywnego zaczyna się kształtować podczas procesu karbonizacji prekursora węgla. Powstają wówczas zespoły skondensowanych pierścieni aromatycznych o różnej wielkości, stanowiące ośrodki tworzenia grafitopodobnych mikrokrystalitów [4].
Według IUPAC (The International Union of Pure and Applied Chemistry) pory klasyfikuje się pod względem rozmiaru na [5]:
- Mikropory, <2nm
- Ultramikropory, <0,5nm
- Supermikropory, od 1 do 2nm
- Mezopory, od 2 do 50nm
- Makropory, >50nm
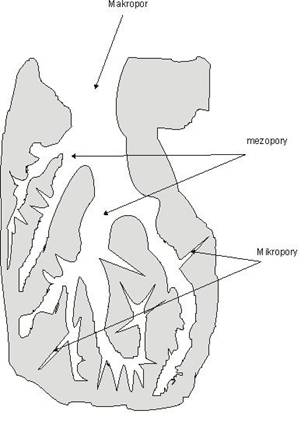
Rysunek 1 Rozkład porów w węglu aktywnym
Największą zdolność adsorpcyjną mają mikropory, ponieważ ich wielkość jest porównywalna z wielkością adsorbowanych cząstek. Mezopory oprócz znaczącego udziału w adsorpcji odgrywają także rolę głównych arterii dla adsorbatu, a makropory pełnią głównie funkcję transportową [2].
- Mikropory
- Charakterystyczną cechą mikroporów jest znacznie większa energia adsorpcji niż w mezo- czy makroporach. Wiąże się to ze zwiększoną zdolnością adsorpcyjną w obszarze małych ciśnień. Poza tym na całej przestrzeni mikroporów istnieje pole sił adsorpcyjnych[6].
- Mezopory
- Ścianki tych porów ukształtowane są przez bardzo duża liczbę atomów lub cząsteczek substancji adsorbentu. Działanie sił adsorpcyjnych przejawia się w niedużej odległości od ścianek, dlatego na powierzchni porów następuje tworzenie kolejnych warstw adsorpcyjnych, kończące się zapełnieniem zgodnie z mechanizmem kondensacji kapilarnej. Mezopory odgrywają rolę w adsorpcji oraz pełnią główną rolę w arterii transportowej dla adsorbatu [7,8].
- Makropory
- Nie odgrywają istotnej roli w procesie adsorpcji. Ich powierzchnia właściwa jest bardzo mała i wynosi od 0,5 do 0,2 m2/g [9].
- Czynnikiem wpływającym na efektywność adsorpcyjną jest rozkład wielkości porów, przykładem tego może być adsorpcja naturalnej materii organicznej (NMO), w skład, której wchodzą głównie wielkocząsteczkowe substancje humusowe i polisacharydy (rys. 2).
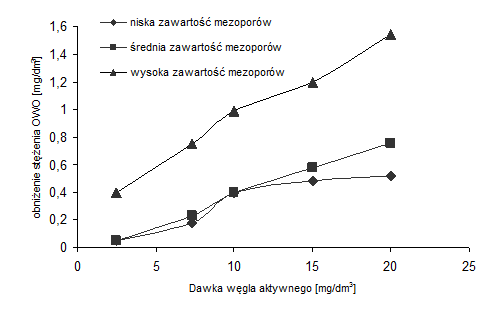
Rysunek 2 Izotermy adsorpcji NMO na węglach aktywnych o różnych zawartościach mezoporów [13]
Ze względu na silną zależność pomiędzy strukturą porowatą i efektywnością adsorpcji różnych adsorbatów, zaleca się dobór odpowiedniego adsorbentu do konkretnego zastosowania (tab.1),
Tabela 1 Zalecane przeznaczenie węgli aktywnych o różnej strukturze porowatej do oczyszczania wody [14]
| Przeznaczenie węgla aktywnego | ||
Usuwanie zanieczyszczeń o małych rozmiarach cząstek | Usuwanie zanieczyszczeń o różnych rozmiarach cząstek | Usuwanie barwy | |
Średnica porów [nm] | Objętość porów [cm3g-1] | ||
<20 | 0,5-0,7 | 0,4-0,36 | 0,3-0,5 |
W wodzie znajdują się zanieczyszczenia o różnej wielości cząsteczek, dlatego też jest wskazanie na stosowanie adsorbentów zawierających pory o szerokim zakresie średnic. [10, 11, 12]
Adsorbenty węglowe są wykorzystywane także do adsorpcji z fazy gazowej w różnych gałęziach przemysłu. Do głównych kierunków wykorzystywania procesu adsorpcji z fazy gazowej na węglach aktywnych można zaliczyć usuwanie z przemysłowych gazów odlotowych substancji szkodliwych dla człowieka i środowiska, odzyskiwanie składników z gazów produkcyjnych, rozdzielanie mieszanin gazowych na składniki [2].
Nowe możliwości stosowania procesu adsorpcji na węglach aktywnych stanowią węglowe sita molekularne. W porównaniu z typowymi węglami aktywnymi, charakteryzują się wąskim rozkładem objętości mikroporów według ich wielkości. Węglowe sita molekularne znalazły wiele zastosowań np. do rozdzielania powietrza na składniki – tlen i azot [2].
1.2 Otrzymywanie węgli aktywnych
Węgiel aktywny jest otrzymywany w procesie karbonizacji i aktywacji materiałów węglowych głównie pochodzenia roślinnego. Podstawowe parametry otrzymywanych węgli aktywnych zależą od rodzaju i właściwości zastosowanego surowca [3] oraz od technologii produkcji.
1.2.1 Surowce do otrzymywania węgli aktywnych
Surowiec przeznaczony do produkcji węgli aktywnych metodą parowo-gazową, powinien mieć: małą zawartość części lotnych, duża zawartość węgla pierwiastkowego, odporność mechaniczną na rozkruszenie i ścieranie.
W celu otrzymywania węgli aktywnych, są stosowane różnorodne materiały pochodzenia organicznego: drewno, torf, lignina, pestki owoców, skorupy orzechów oraz węgle kopalne, rzadziej materiały pochodzenia syntetycznego [2].
1.2.2 Karbonizacja
Pierwszym etapem otrzymywania adsorbentów węglowych jest karbonizacja, podczas której powstaje tzw. pierwotna struktura porowata węgla. Proces karbonizacji przebiega w temperaturze 500-1000oC, w atmosferze gazu inertnego, powstający karbonizat nabiera mechanicznej wytrzymałości, przy czym w wyniku wydzielania lotnych substancji materiał wzbogaca się w węgiel.
Właściwości karbonizatu zależą od warunków pirolizy, a wśród nich takich parametrów jak [2]:
- Końcowa temperatura karbonizacji - im wyższa jest temperatura tym głębiej przebiegają procesy pirolizy materiałów (z reguły powstają wytrzymalsze mechanicznie granule).
- Czas karbonizacji - czas przetrzymywania węgla w końcowej temperaturze karbonizacji wpływa na większe uporządkowanie budowy zwartej substancji węglowej.
- Szybkość osiągnięcia temperatury końcowej - gdy wzrost temperatury jest szybki, wówczas poszczególne fazy rozkładu termicznego prekursora węgla oraz wtórne reakcje produktów pirolizy między sobą nakładają się, a przez to powstanie struktury porowatej w karbonizacje jest trudniejsze do kontrolowania. Duża szybkość wzrostu temperatury nie sprzyja również większemu uporządkowaniu chemicznej budowy substancji węglowej. W przypadku szybkiego ogrzewania, z karbonizowanego surowca wydziela się duża ilość części lotnych. W efekcie tego powstają zazwyczaj pory o większych rozmiarach.
- Atmosfera, w której odbywa się karbonizacja - wpływa również na rozkład termiczny węgla oraz na przebieg wtórnych reakcji produktów pirolizy między sobą i reakcji tychże ze stałym karbonizatem.
Ilość uzyskanego karbonizatu jest mniejsza, za to jego reaktywność jest większa, gdy wydzielające się podczas pirolizy gazy lub pary są szybko usuwane za pomocą przepływającego gazu obojętnego lub gazów spalinowych.
1.2.3 Aktywacja
Produkt karbonizacji ma słabo rozwiniętą strukturę porowatą i z reguły wykazuje niewielką powierzchnię właściwą. Dlatego też w celu uzyskania materiałów porowatych konieczny jest proces aktywacji. Procesy otrzymywania materiałów węglowych o rozwiniętej porowatości i powierzchni właściwej otrzymuje się na drodze aktywacja fizycznej (gazowa) lub chemicznej [2].
Aktywację fizyczną prowadzi się w atmosferze [2]:
- Pary wodnej (800-1000oC)
- Dwutlenku węgla (800-1000oC)
- Kombinacją wyżej wymienionych gazów
- Tlenu (rzadziej stosowany)
Znaczny wpływ na stopień rozwinięcia struktury porowatej podczas aktywacji fizycznej mają temperatura procesu, reaktywność materiału węglowego oraz aktywność czynnika zgazowującego, bowiem limitują one szybkość aktywacji. Z wymienionych czynników utleniających najbardziej aktywny jest tlen, a najmniej dwutlenek węgla. Z reguły stosowanie dwutlenku węgla w procesie aktywacji wpływa na silniejsze rozwinięcie struktury mikroporowatej, a pary wodnej – struktury porowatej o większym udziale mezoporów.
Ponadto, przy odpowiednio dobranym surowcu, materiał aktywny zachowuje kształt ziaren prekursora, np. z skorup orzecha kokosowego można otrzymać granulat węgla aktywnego o bardzo małej ścieralności, a aktywacja materiałów włóknistych prowadzi do wytworzenia tkanin lub mat o zdolnościach sorpcyjnych.
Przy pomocy aktywacji fizycznej otrzymuje się węgle aktywne o powierzchni właściwej 600-1000 m2/g. W trakcie procesu występuje duży ubytek masy surowca – nawet powyżej 70% [2]. Główną zaletą aktywacji fizycznej jest niski koszt wytworzenia jednostkowej ilości węgla aktywnego.
Aktywacja chemiczna jest procesem jednoetapowym, łączący karbonizację i aktywację w jednym etapie, polegającym na reakcji prekursora węgla (torf, drewno, łupiny orzechów, pestki owoców) z czynnikiem aktywującym, którym w skali przemysłowej jest H3PO4 lub ZnCl2. Temperatura procesu ok. 600oC.
W porównaniu z aktywacją fizyczną, aktywacja chemiczna pozwala na [3]:
- Ominięcie wstępnego etapu karbonizacji surowca,
- Otrzymanie węgla aktywnego z większą wydajnością i o silniej rozwiniętej strukturze porowatej,
- Uzyskaniu materiału węglowego o bardzo dużej powierzchni właściwej,
- Obniżenie temperatury procesu,
- Ograniczenie tworzenia się smoły,
- Odpopielanie i odsiarczenie materiału węglowego.
Przeprowadzenie procesu aktywacji chemicznej jest mniej skomplikowane niż aktywacji fizycznej, a jednocześnie stwarza więcej możliwości w procesie tworzenia węgli aktywnych o wymaganym rozkładzie szerokości porów. Po zakończeniu termicznego etapu procesu, konieczne jest jednak oddzielenie stałych reagentów i produktów ich rozkładu od materiału porowatego oraz ich regeneracja lub utylizacja.
To właśnie ten etap aktywacji chemicznej silnie wpływa na wyskoki koszt procesu oraz jest na tyle kłopotliwy, że ta metoda aktywacji jest stosowana rzadziej niż aktywacja fizyczna [3]. Technologie aktywacji przy użyciu chlorku cynku zostały całkowicie wycofane ze względu na ochronę środowiska naturalnego.
2. Cel i zakres pracy
Praca dotyczy otrzymywania adsorbentów węglowych z surowca syntetycznego (PET) z użyciem związków nieorganicznych. Celem było uzyskanie jednoetapowej metody, polegającej na karbonizacji prekursora węgla z dodatkiem substancji nieorganicznej, z pominięciem klasycznego etapu aktywacji fizycznej. W tym celu należało użyć takiego związku nieorganicznego, który będzie ulegał rozkładowi termicznemu z wydzieleniem produktów gazowych będących typowymi czynnikami aktywującymi stosowanymi w procesie aktywacji fizycznej (CO2, para wodna), a przy tym powodujący tworzenie się struktury porowatej węgla.
Opracowanie metody, która pozwalałaby na kontrolę zawartości mikroporów i mezoporów w węglach aktywnych jest ważna ze względu na wykorzystanie adsorbentów do selektywnej adsorpcji. Istotnym jest także ich wykorzystanie do oczyszczania wód zawierających zanieczyszczenia o różnych wielkościach cząstek, a także do preparatyki katalizatorów na bazie węgli aktywnych. Dlatego też dodatkowym celem było opracowanie sposobu kontroli struktury porowatej węgla, poprzez generowanie mikroporów (działanie wydzielających się CO2/para wodna na materiał węglowy) oraz tworzenie mezoporów (działanie stałego produktu rozkładu – tlenku na materiał węglowy).
Odpowiedni dobór jakościowy i ilościowy reagentów może być sposobem na kontrolę struktury porowatej otrzymywanych adsorbentów, co jest bardzo ważne z punktu widzenia otrzymywania adsorbentu o określonych, powtarzalnych właściwościach. Oprócz tego, z racji narastających problemów związanych z zanieczyszczeniem środowiska naturalnego, otrzymanie materiałów służących ochronie środowiska z surowców będących zanieczyszczeniem, jest bardzo ważnym aspektem.
3. Charakterystyka surowców i odczynników
3.1 Poli(tereftalan etylenu) - (PET)
Polimer z grupy poliestrów, który otrzymywany jest na drodze polikondensacji z tereftalanu dimetylowego (DMT) i glikolu etylenowego (GE) [56].
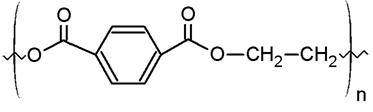
Rysunek 8 - Łańcuch PET [56]
PET otrzymywany w wyniku szybkiego ochłodzenia (produktu polikondensacji) jest amorficzny i ma gęstość 1,33 g/cm3. Stosunkowo łatwo (szczególnie w podwyższonej temperaturze) ulega krystalizacji. Gęstość krystalicznego PET wynosi 1,45 g/cm3. Temperatura topnienia zależy od stopnia polimeryzacji i wynosi 255-264oC [57].
Na podstawie doniesień literaturowych stwierdzono brak emisji dymu podczas rozkładu termicznego PET w niższym zakresie temperatur 200-300oC. Natomiast w temperaturach wyższych 400-700°C proces rozkładu przebiegał z wydzieleniem dużej ilości trudno lotnych związków, tworzących jasnożółty dym. W skład emitowanego dymu wchodzą: złożone mieszaniny związków aromatycznych, na które składa się kwas tereftalowy i jego estry, kwas benzoesowy, para-podstawione kwasy benzoesowe, kwas 2-naftoesowy,1, 4-diacetylobenzen, 4-acetylobifenyl, oligomery PET, śladowe ilości wielopierścieniowych węglowodorów aromatycznych i ftalanu di butylu [65].
W badaniach zastosowano PET produkowany przez firmę Elana S.A
6. Omówienie wyników i dyskusja
6.1 Analiza XRD
Analizie XRD poddano surową mieszaninę 3MgCO3·Mg(OH)2·3H2O/PET oraz mieszaninę 3MgCO3·Mg(OH)2·3H2O z PET karbonizowaną w 850oC, w różnych okresach czasu (1h, 2h). Dyfraktogramy materiałów węglowych przedstawiono na rysunkach 14, 15.
Dyfraktogram mieszaniny wyjściowej 3MgCO3·Mg(OH)2·3H2O/PET (rys. 14) nie wykazuje wyraźnych pików, które mogłyby wskazywać na obecność faz krystalitów, prawdopodobnie jest to związane z amorficznym stanem pośredniego produktu rozkładu 3MgCO3·Mg(OH)2·3H2O, którym jest MgCO3·Mg(OH)2 ( rozdz. 6.3)
Rysunek 14 Dyfraktogram mieszaniny wyjściowej 3MgCO3•Mg (OH)2•3H2O:PET
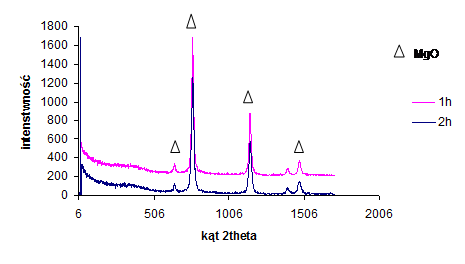
Rysunek 15 Dyfraktogram próbki otrzymanej z 3MgCO3•Mg (OH)2•3H2O/PET (50:50), czas karbonizacji 1h, 2h
Analiza składu fazowego powstałych materiałów węglowych na bazie 3MgCO3·Mg(OH)2·3H2O i PET (rys. 15) wykazała istnienie nowej fazy krystalicznej w postaci MgO, po ogrzaniu do 850oC.
Na podstawie dyfraktogramu (rys. 15) obliczono wielkość krystalitów MgO w funkcji czasu (tab. 6).
Tabela 6 Wpływ wielkości krystalitu na porowatość węgla
Próbka | Temperatura karbonizacji [oC] | Czas karbonizacji [h] | Wielkość krystalitu [nm] |
3MgCO3·Mg(OH)2·3H2O:PET | 850 | 1h | 13 |
2h | 10 |
Jak widać czas karbonizacji nie ma istotnego wpływu na wielkość krystalitów MgO powstających w preparowanych próbkach.
6.4 Pomiar izoterm adsorpcji, desorpcji azotu w temperaturze 77K
Izotermę mieszaniny 3MgCO3·Mg(OH)2·3H2O z PET (karbonizacja 850oC, w czasie 1h) przedstawiono na rysunku 22.
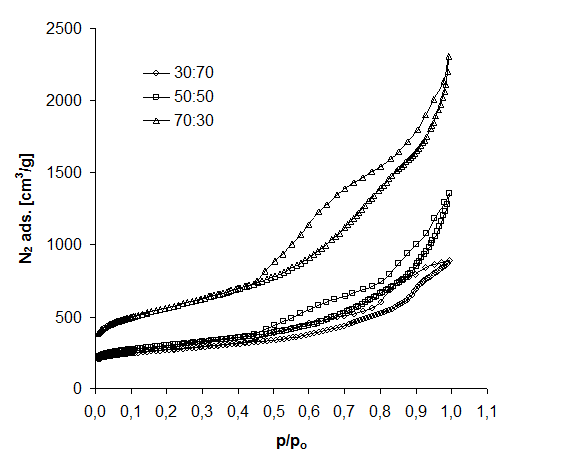
Rysunek 22 Izotermy adsorpcji azotu 3MgCO3*Mg(OH)2*H2O/PET
Zauważa się tendencję wzrostową adsorpcji azotu wraz z zwiększeniem zawartości 3MgCO3·Mg(OH)2·3H2O w mieszaninie wyjściowej. Otrzymane izotermy wskazują na obecność mezoporów. Zwiększona adsorpcja azotu może mieć związek z porowatością materiału, która może mieć związek z wydzielaniem się podczas procesu ogrzewania 3MgCO3·Mg(OH)2·3H2O produktów gazowych takich jak CO2 i H2O (rozdz. 6.3).
Zbliżoną zbliżony kształt izotermy względem próbki 3MgCO3·Mg(OH)2·3H2O:PET 70:30 (rys. 22) wykazuje mieszanina Mg(OH)2 z PET 50:50 (rys. 23). Widoczna pętla histerezy wskazuje na obecność mezoporów co może mieć związek z wydzieleniem się podczas procesu ogrzewania Mg(OH)2 wody (rozdz. 6.3)
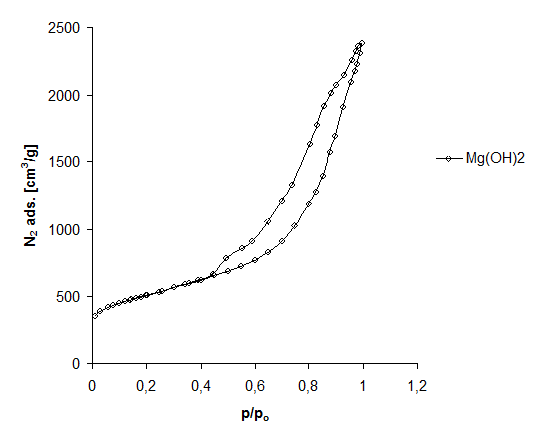
Rysunek 23 Izoterma adsorpcji azotu Mg(OH)2:PET 50:50
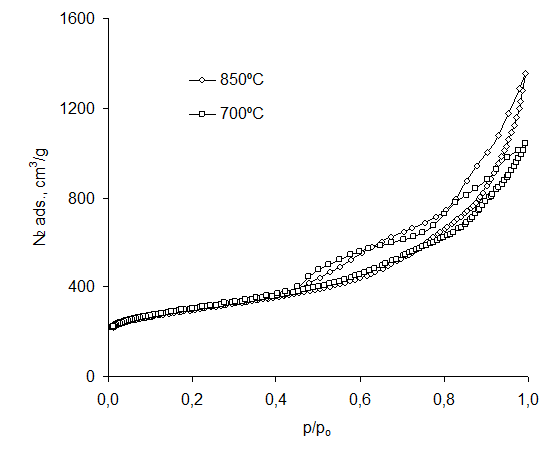
Rysunek 24 Izoterma adsorpcji azotu MgO:PET 50:50
Otrzymane izotermy MgO/PET 50:50 charakteryzują się niewielką tendencję wzrostową adsorpcji azotu wraz ze wzrostem temperatury karbonizacji. Pętla histerezy wskazuje na obecność mezoporów.
Wartości powierzchni właściwej oraz mikro- i mezoporów (tab. 13) uzyskano w oparciu o izotermy (rys. 22, 23, 24).
Tabela 13 Wartość powierzchni właściwej BET i względny rozkład udział porów w badanych próbkach
Lp. | Próbka (skład, stosunek ilościowy, temp. Karbonizacji) | Powierzchnia właściwa BET [m2/g] | Powierzchnia mezoporów [m2/g] | Udział mezoporów BJH/BET [%] |
1 | 3MgCO3·Mg(OH)2·3H2O:PET 30:70, 850oC | 944 | 470 | 49 |
2 | 3MgCO3·Mg(OH)2·3H2O:PET, 50:50, 850oC | 1049 | 638 | 61 |
3 | 3MgCO3·Mg(OH)2·3H2O:PET, 70:30, 850oC | 1984 | 1426 | 72 |
5 | MgO:PET, 50:50, 700oC | 800 | 338 | 42 |
6 | MgO:PET, 50:50, 850oC | 790 | 341 | 43 |
7 | Mg(OH)2:PET 50:50, 850oC | 1794 | 1517 | 76 |
Zauważa się tendencję wzrostową powierzchni właściwej i udziałów mezoporów mieszaniny 3MgCO3·Mg(OH)2·3H2O z PET wraz ze wzrostem zawartości związku magnezu, przy stosunkowo niewielkich zmianach udziału mikroporów. Wysoką powierzchnię właściwą i powierzchnię mezoporów uzyskano także w mieszaninie Mg(OH)2 z PET. Zbadano wpływ temperatury karbonizacji na próbkę MgO/PET, otrzymane powierzchnie właściwe i powierzchnie mezoporów w próbkach karbonizowanych w temperaturze 700oC i 850oC są porównywalne.
7. Próby oszacowania przydatności otrzymanych materiałów węglowych do procesów adsorpcji
Z faktu obecności mikro- i mezoporów w otrzymanych materiałach węglowych przeprowadzono adsorpcję na modelowych związkach o różnych masach cząsteczkowych i wielkościach cząsteczek takich jak: fenol, czerwień reaktywna, kwasy humusowe.
7.3.2 Pomiar stężenia kwasu humusowego na podstawie pomiaru zawartości ogólnego węgla organicznego
W celu określenia przydatności otrzymanych materiałów węglowych do usuwania związków o większej wielkości cząstek, przeprowadzono badania adsorpcji kwasów humusowych na węglach aktywnych otrzymanych z: 3MgCO3·Mg(OH)2·3H2O/PET o następujących proporcjach wagowych 30:70, 50:50, 70:30 oraz Mg(OH)2/PET 50:50. Materiały węglowe były otrzymane w 850oC, w czasie ogrzewania 1h.
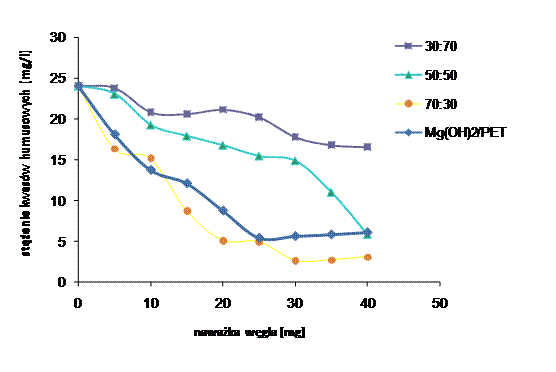
Rysunek 34 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (30:70, 50:50, 70:30), Mg(OH)2/PET 50:50
Adsorpcja kwasów humusowych zwiększa się z zawartością związku magnezu w mieszaninie wyjściowej (rys. 34). Jak wynika z danych przedstawionych w rozdziale 6.4, węgle porowate otrzymane z mieszaniny wyjściowej o większym udziale wagowym 3MgCO3·Mg(OH)2·3H2O, zawierały więcej mezoporów. Materiał węglowy otrzymany z Mg(OH)2/PET 50:50, wykazał się zbliżoną zdolnością adsorpcyjną kwasów humusowych do 3MgCO3·Mg(OH)2·3H2O/PET 70:30 , wynika to ze zbliżonej powierzchni mezoporów (rozdz. 6.4).
Zbadano adsorpcję kwasów humusowych na węglach aktywnych otrzymanych z mieszaniny wyjściowej 3MgCO3·Mg(OH)2·3H2O/PET 50:50, karbonizowanej w 850°C, w różnych okresach czasu (0,5h, 1h, 2h).
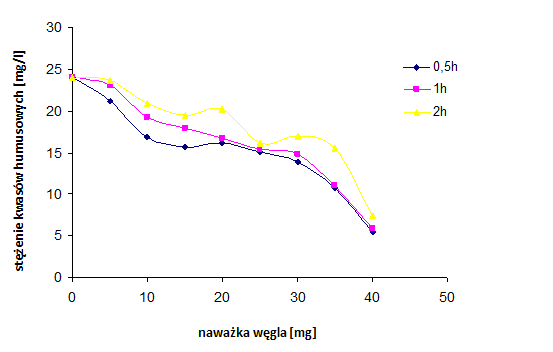
Rysunek 35 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (różne czasy karbonizacji)
Rysunku 35 pokazuje, że czas karbonizacji nie wpływa znacząco na zdolność adsorpcyjną kwasów humusowych. Może to wynikać, ze zbliżonej powierzchni mezoporów otrzymanych węgli aktywnych.
Przeprowadzono proces adsorpcji kwasów humusowych na węglach aktywnych otrzymanych z mieszaniny wyjściowej MgO/PET 50:50, karbonizowanej w 700oC i 850oC, w czasie 1 h (rys. 36)
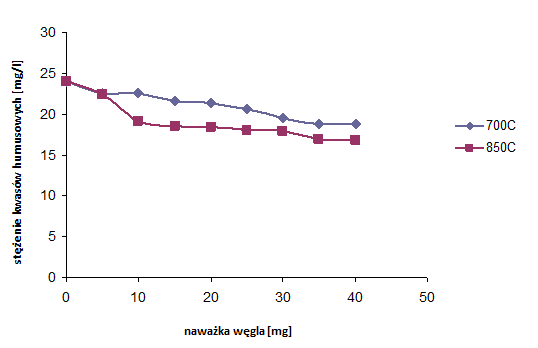
Rysunek 36 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny MgO/PET o stosunku wagowym 50:50 (różne temperatury karbonizacji)
Niewielki wzrost adsorpcji kwasów humusowych zauważa się na próbce karbonizowanej w 850oC. Materiał węglowy otrzymany w wyższej temperaturze wykazuje się nieznacznie większą powierzchnią mezoporów(rozdz. 6.4).
8. Wnioski
- Zwęglanie PET w mieszaninie ze związkami magnezu badanymi w pracy pozwala na uzyskanie porowatych materiałów węglowych.
- Niezależnie od zastosowanego związku magnezu w mieszaninie z PET, po zakończonym procesie karbonizacji utworzyła się nowa faza krystaliczna w postaci MgO.
- Struktura porowata otrzymanych adsorbentów węglowych zależy od rodzaju zastosowanego związku magnezu.
- Powierzchnia BET i mezoporów rosną wraz ze wzrostem zawartości zasadowego węglanu magnezu w mieszaninie wyjściowej.
- Efektywność adsorpcji fenolu rosła wraz ze wzrostem powierzchni właściwej i udziałem mikroporów w badanych adsorbentach węglowych.
- Efektywność adsorpcji czerwieni reaktywnej, kwasów humusowych rosła wraz ze wzrostem powierzchni właściwej i udziałem mezoporów w badanych adsorbentach węglowych.
- Czas karbonizacji nie ma wpływu na właściwości adsorpcyjne otrzymanych węgli aktywnych, wobec zastosowanych adsorbatów.
- Wzrost temperatury karbonizacji powoduje zwiększenie się zdolności adsorpcyjnej na otrzymanych materiałach węglowych, wobec stosowanych adsorbatów.
Bibliografia
1. K. Nakagawa, A. Namba, S.R. Mukai, H. Tamon, P. Ariyadejwanich, W. Tanthapanichakoon, Water Res. 38, 1791–1798, 2004
2. H. Jasińska, A. Świątkowski, J. Choma, Węgiel aktywny, WNT, Warszawa 1985
3. H. Marsh, E.A. Heintz, F. Rodriguez-Reinoso, Activated Carbon: Structure characterization, preparation and applications, w Introduction to Carbon Technologies, 1997
4. Satish M Manocha, Porus Carbon, 335-348, India, 2003
5. K.S.W. Sing, D.H. Everett, R.A.W. Haul, L. Moscou, P.A. Pierotti, J.Rouquerol, Pure Appl. Chem. 57, 603, 1985
6. A.M. Anielak, Wybrane zagadnienia z technologii ścieków przemysłowych, Koszalin 1999
7. W. Adamski, Modele pracy biologicznej aktywnych złóż sorpcyjnych w oczyszczaniu wody, WKUP, Koszalin 1997
8. N. Kielce, Podstawy techniki adsorpcyjnej, WNT, Warszawa 1998
9. B.J. Stiepanow, Podstawy chemii i technologii barwników organicznych, WNT, Warszawa 1968
10. G.L. Culp, R.L. Cupl: New concepts in water purification, Reinhold, New York 1974
11. P. Matson, H.B. Mark: Activated carbon: surface chemistry and adsorption from solution, Marcel Dekker, New York 1971
12. D.M. Ruthven: Principles of adsorption and adsorption processes, Wiley, New York 1984
13. G. Newcombe, J. Morrison, C. Hepplewhite, D. Knappe: Wat. Sci. Technol.: Water Supply 2(2), 59, 2002
14. Ullmann's Encyclopedia of Indriustrial Chemistry 6th Edition, vol. 6, Wiley-VCH, Weinheim
15. M. Inagaki, New carbons Control of structure and functions, Elservier Amsterdam-Lausanne-New York-Oxford-Shannon-Singapore-Tokyo, 2000
16 . H. Juntgen, K. Knoblauch, H. Munzner, Chem-Ing-Techn 45:533–7, 1973
17. K. Miura Shokubai (Catalysts Catalysis) 41:25–30, 1999
18. K. Miura, J. Hayashi, K. Hashimoto, Carbon 29:653–60, 1991
19. K. Miura, J. Hayashi, K. Hashimoto, Carbon 30:946–7, 1990
20. K. Miura, H. Nakagawa, T. Kawano, Tanso 185:249–55, 1998
21. H. Nakagawa, K. Watanabe, Y. Harada, K. Miura, Carbon 37(9):1455- 1461, 1999
22. T. Kyotani, Control of pore structure in carbon, Carbon 38:269–286, 2000
23. Y. Kawabuchi, S. Kawano, I. Mochida, Carbon 34:711–7, 1996
24. Y. Kawabuchi, H. Oka, S. Kawano, I. Mochida, N. Yoshizawa, Carbon 36(4):377–82, 1998
25. S. K. Verma, Y. Nakayama, P.L. Walker Jr., Carbon 31:533–4, 1993
26. K. Laszlo, Microporous and Mesoporous Materials 80 ,Budapest, Hungary, 205-211, 2005
27. H. Marsh, B. Rand, Carbon 9:63–77, 1971
28. J.J. Freeman, F.G.R. Gimblett, R.A. Roberts, K.S.W. Sing, Carbon 1988;26:7–29. A.Tomita, Y. Yuhki, K. Higashiyama, T. Takarada, Y. Tamai, N.Kyokaishi (J Fuel Soc Jpn) 64:402–8, 1985
30. K. Shimazaki, J. Chem. Soc. Jpn., 807–12, 1993
31. A.Oya, S. Yoshida, J. Alcaniz-Monge, A. Linares-Solano, Carbon33, 1085–90, 1995
32. H. Tamai, T. Kakii, Y. Hirota, T. Kumamoto, H. Yasuda, Chem Mater 8:454–62, 1996
33. T. Bandosz, J. Jagiello, K. Putyera, J.A .Schwarz. Chem Mater 8:2023, 1996
34. Błędzik A.K., Recykling materiałów polimerowych, Wydawnictwo Naukowo Techniczne, Warszawa 1997
35. Błędzik A.K., Kardosz D., Opakowanie, Zeszyt Specjalny nr. III, 1995,
36. Szlezyngier W., Tworzywa sztuczne, Tom II, Oficyna Wydawnicza Politechniki Rzeszowskiej, Rzeszów, 1996
37. M. Blazso, Journal of Analytic and Applied Pyrolysis 39 1, 1997
38. K. Laszlo , A. Bota , I. Dekany, Carbon 41, 1205–1214, 2003
39. K. Laszlo , E. Tombacz , K. Josepovits, Carbon 39, 1217–1228, 2001
40. F. Zhang, H. Itoh, J. Hazard. Mater. 2003;B101:323–337, 2003
41. M. Inagaki, S. Kobayashi, F. Kojin, N. Tanaka, T. Morishita, B. Tryba, Carbon 42, 3153–3158, 2004
42. Laszko K., Bota A., Nagy L.G., Conference of Collodi Chem., Proc., 7th, 124-127, 1996
43. Bota A., Laszko K., Nagy L.G., Langmuir, 13(24), 6502-6509, 1997
44. Laszko K., Bota A., Nagy L.G., Canasso I., Physicochemical and Enginneering Aspects, 151 (1-2), 311-320, 1999
45. Gryta M., Morawski A. W., Karbo-Energochemia-Ekologia,, 43(5), 167-170, 1998
46. Gryta M., Morawski A.W., III Ogólnopolska Konferencja Naukowa, Inżynieria Procesowa w ochronie środowiska, Materiały konferencyjne, 306-310, 1997
47. C. Barriocanal, M.A. Diez, R. Alvarez, PET recycling for the modification of precursors in carbon materials manufacture, 45-51, 2005
48. I. Fernandez-Morales, M..C. Almazan-Almazan, M. Perez-Mendoza, M.Domingo-Garcia, F.J. Lopez-Garzon, PET as precursor of microporous carbons: preparation and characterization, 107-115, 2005
49. M.T. Kartel, N.V. Sych, M.M. Tryba, V.V. Strelko , Preparation of porous carbons by chemical activation of polyethyleneterephthalate, 1013-1024, 2005
50. M.T. Kartel, N.V. Sych, M.M. Tsyba, V.V. Strelko, Carbon 44, 1013–1024, 2006
51. K. Nakagawa, S. R. Mukai, T. Suzuki, H. Tamon, Carbon 41:823–831, 2003
52. M.C. Almazan-Almazan, M. Preze-Mendoza, F.J. Lopez-Domingo, I.Fernandez-Morales, M. Domingo-Garcia, F.J. Lopez-Garzon, Microporous an Mesoporous Materials 106, 219-228, 2007
53. J.B. Parra, C.O. Ania, A. Arenillas, F. Rubeira, J.J. Pis, International Conference on Carbon, Oviedo, Spain, 152–155 (extended abstracts), 2003.
54. T. Morishita, Y. Soneda., T. Tsumura, M. Inagaki, Carbon 44, 2360-2367, 2006
55. M. Inagaki, S. Kobayashi, F. Kojin, N. Tanaka, T. Morishita, B. Tryba, Carbon 42, 3153-3158, 2004
56. http://pl.wikipedia.org/wiki/Poli(tereftalan_etylenu), 09.02.2008 r.
57.http://www.wiedzainfo.pl/wyklady/124/pet_czyli_poli_tereftalan_etylenu_produkcja_zastosowanie_recykling.html, 09.02.2008 r.
58. N. Khana, D. Dollimore, K. Alexander, F.W. Wilburn, The origin of the exothermic peak in the thermal decomposition of basic magnesium carbonate, 321-33, 2001
59. G. Helou, S.A. Tariq, The pyrolysis of basic magnesium carbonate trihydrate, 123-126, 1993
60. http://pl.wikipedia.org/wiki/Wodorotlenek_magnezu 16.03.2008
61. E. M.van Merwe, C. A. Strydom, Quantitative thermogravimetric analysis of binary mixtures Magnesium hydroxide and magnesium acetate,149-156, 2004
62. http://pl.wikipedia.org/wiki/Tlenek_magnezu, 09.02.2008 r.
63. http://pl.wikipedia.org/wiki/Fenol, 09.02.2008 r.
64. http://pl.wikipedia.org/wiki/Kwasy_humusowe 12.03.2008
65. Dzięcioł, M., Baran, J. GC-MS analysis of smoke composition during thermal degradation of poly(ethylene terephthalate), 669-676, 2001
Spis rysunków
Rysunek 1 Rozkład porów w węglu aktywnym
Rysunek 2 Izotermy adsorpcji NMO na węglach aktywnych o różnych zawartościach mezoporów [13]
Rysunek 3 - Krzywe rozkładu wielkości porów w molekularnym sicie węglowym przygotowanym z mieszaniny węgla i smoły węglowej [26]
Rysunek 4 Kontrola struktury porowatej materiałów polimerowych metodą "blending" [15]
Rysunek 6 – Zależność ubytku masy od czasu aktywacji [49]
Rysunek 7 Wpływ temperatury na ubytek masy i porowatość węgla otrzymanego z chemicznej aktywacji[49]
Rysunek 8 - Łańcuch PET [56]
Rysunek 9 Schemat wygrzewania próbki
Rysunek 10 Krzywa wzorcowa czerwieni reaktywnej
Rysunek 11 Krzywa wzorcowa fenolu
Rysunek 12 Krzywa wzorcowa dla roztworu kwasów humusowych
Rysunek 13 Krzywa wzorcowa dla roztworu kwasów humusowych
Rysunek 14 Dyfraktogram mieszaniny wyjściowej 3MgCO3•Mg (OH)2•3H2O:PET
Rysunek 15 Dyfraktogram próbki otrzymanej z 3MgCO3•Mg (OH)2•3H2O:PET (50:50), czas karbonizacji 1h, 2h
Rysunek 16 Próbka 3MgCO3*Mg(OH)2*H2O:PET 50:50, karbonizowana w 850oC, czas karbonizacji: a) 1h, b)2h
Rysunek 17 Termogram dla czystej próbki PET
Rysunek 18 Termogram dla czystego 3MgCO3*Mg(OH)2*3H2O
Rysunek 19 Termogram dla czystego Mg(OH)2
Rysunek 20 Termogramy dla próbek 3MgCO3*Mg(OH)2*3H2O/PET (różne proporcje wagowe)
Rysunek 21 Termogram dla próbki Mg(OH)2:PET o stosunku wagowym 50:50
Rysunek 22 Izotermy adsorpcji azotu 3MgCO3*Mg(OH)2*H2O/PET
Rysunek 23 Izoterma adsorpcji azotu Mg(OH)2:PET 50:50
Rysunek 24 Izoterma adsorpcji azotu MgO:PET 50:50
Rysunek 25 Adsorpcja fenolu na węglach aktywnych otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (30:70, 50:50, 70:30), Mg(OH)2:/PET 50:50
Rysunek 26 Adsorpcja fenolu na węglach aktywnych (różne czasy karbonizacji) otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O:PET 50:50
Rysunek 27 Adsorpcja fenolu na węglach aktywnych (wpływ temperatury karbonizacji) otrzymanych z mieszaniny MgO:PET 50:50.
Rysunek 28 Adsorpcja czerwieni reaktywnej na węglach aktywnych (różne proporcja wagowe) otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O:PET
Rysunek 29 Adsorpcja czerwieni reaktywnej na węglach aktywnych (różne czasy karbonizacji) otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O:PET 50:50
Rysunek 30 Adsorpcja czerwieni reaktywnej na węglach aktywnych (wpływ temperatury karbonizacji) otrzymanych z mieszaniny MgO:PET 50:50
Rysunek 31 Adsorpcja kwasów humusowych na węglach aktywnych otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (30:70, 50:50, 70:30), Mg(OH)2/PET 50:50
Rysunek 32 Adsorpcja kwasów humusowych na węglach aktywnych otrzymanych z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (różne czasy karbonizacji)
Rysunek 33 Adsorpcja kwasów humusowych na węglach aktywnych otrzymanych z mieszaniny MgO/PET (wpływ temperatury karbonizacji)
Rysunek 34 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (30:70, 50:50, 70:30), Mg(OH)2/PET 50:50
Rysunek 35 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET (różne czasy karbonizacji)
Rysunek 36 Adsorpcja kwasów humusowych na węglach aktywnych, na podstawie pomiaru zawartości ogólnego węgla organicznego z mieszaniny MgO/PET o stosunku wagowym 50:50 (różne temperatury karbonizacji)
Spis tabel
Tabela 1 Zalecane przeznaczenie węgli aktywnych o różnej strukturze porowatej do oczyszczania wody [14]
Tabela 2 Podstawowe właściwości fizykochemiczne (dane producenta)
Tabela 3 Ilościowy skład mieszanin wyjściowych stosowanych do badań
Tabela 4 Przykładowy program temperaturowy
Tabela 5 Warunki karbonizowanych próbek
Tabela 6 Wpływ wielkości krystalitu na porowatość węgla
Tabela 7 Próbki poddane pomiarom TG
Tabela 8 Zawartość procentowa węgla uzyskana z PET
Tabela 9 Warunki karbonizacji oraz ubytki masy surowej próbki PET
Tabela 10 Warunki karbonizacji oraz ubytki masy mieszaniny 3MgCO3*Mg(OH)2*3H2O/PET
Tabela 11 Warunki karbonizacji oraz ubytki masy mieszaniny Mg(OH)2/PET
Tabela 12 Warunki karbonizacji oraz ubytki masy mieszaniny MgO/PET
Tabela 13 Wartość powierzchni właściwej BET i względny rozkład udział porów w badanych próbkach